一、受体动力学
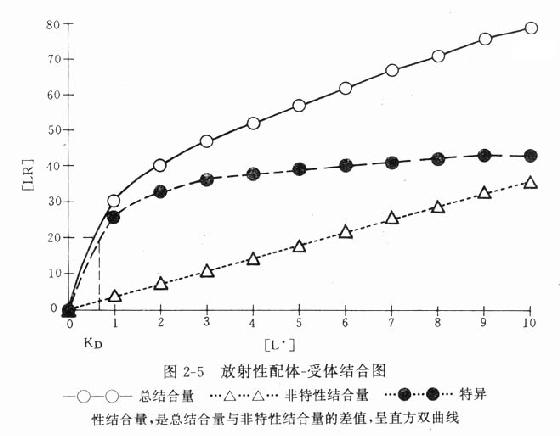
受体动力学一般用放射性同位素标记的配体(L)与受体(R)做结合试验研究。取一定量组织,磨成细胞匀浆,分组加入不同浓度的放射性同位素标记的配体(药物),温孵待反应达平衡后,迅速过滤或离心分出细胞,用缓冲液洗去尚未结合的放射性配体,测定标本的放射强度,这是药物与细胞结合的总量,此后用过量冷配体(未用同位素标记的配体)洗脱特异性与受体结合的放射性配体再测放射强度,这是药物非特性结合量。将总结合量减去非特性结合量就可以获得L-R结合(B)曲线。如果L只与单一R可逆性结合,以B为纵座标,[L]为横座标,L-R结合曲线为直方双曲线(图2-5)。如将横座标改用log[L]([]表示摩尔浓度)则呈典型的S形量效曲线。
按质量作用定律
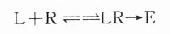
(E代表效应)
反应达到平衡时
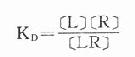
(KD是解离常数)
因为[RT]=[R]+[LR](RT为受体总量),代入上式并经推导得
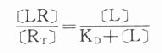
由于只有LR才发挥效应,故效应的相对强弱与LR相对结合量成比例,即
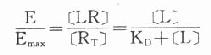
按此公式以E为纵座标,log[L]为横座标作图,结果与实验数据图形完全一致。
当[L]=0时,效应为0,
当[L]>>KD时,[LR]/[RT]=100%,达最大效能,即[LR]max=[RT]。
当[LR]/[RT]=50%时,即EC50时,KD=[L]。
KD表示L与R的亲和力(affinity),单位为摩尔。各药(L)与R亲和力不同,KD越大时亲和力越小,二者成反比。令pD2=-logKD则其值不必用摩尔单位、数值变小且与亲和力成正比,在半对数座标上也较易理解,故pD2较为常用。
药物与受体结合产生效应不仅要有亲和力,还要有内在活性(intrinsicactivity),后者用α表示,0≤α≤100%。故上述公式应加入这一参数:E/Emax=α[LR]/[RT]。两药亲和力相等时其效应强度取决于内在活性强弱,当内在活性相等时则取决于亲和力大小(图2-6)。
将上述受体动力学基本公式([LR]/[RT]=[L]/KD+[L])加以推导改变可将S形量效曲线改变为直线关系,使计算方便很多也准确很多:
1.双倒数图 将上述基本公式两侧取倒数后加以推导得1/[LR]=KD/[L][RT]+1/[RT]。以1/[LR]为纵座标、1/[L]为横座标作图得直线(图2-7),斜率为KD[RT],即KD/Emax,与纵座标交点为1/[RT],即1/Emax,与横座标交点为-1/KD。
2.Scatchard图 推导得公式[LR]/[L]=[RT]/KD-[LR]/KD以[LR]/[L],为纵座标,[LR]为横座标作图也呈直线(图2-8),斜率为-1/[KD],与纵座标交点为[RT]/KD,与横座标交点为[RT]。
这些直线关系图解在受体研究中有重要用途,也可加深对受体动力学的理解
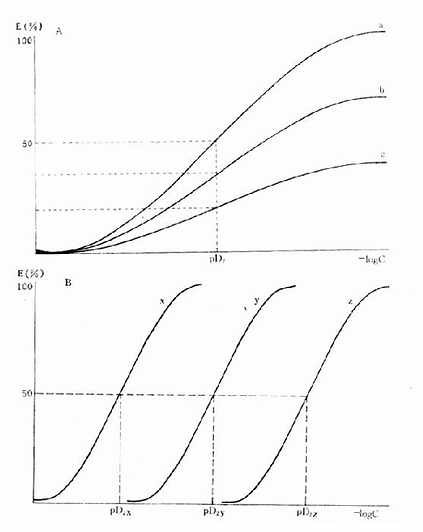
图2-6 药物与受体的亲和力及其内在活性对量效曲线的影响
A图 a,b,c三药与受体的亲和力(pD2)相等,但内在活性(Emax)不等
B图 a,b,c 三药与受体的亲和力(pD2)不等,但内在活性(Emax)相等
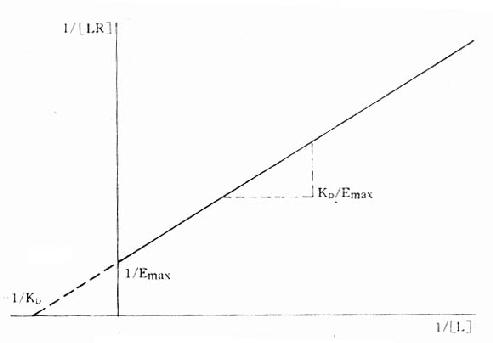
图2-7 受体结合量效关系的双倒数作图
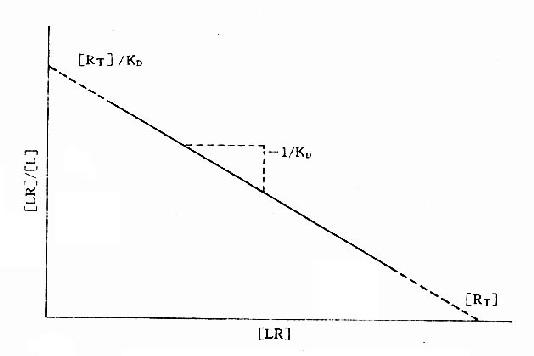
图2-8 受体结合量效关系的Scatchard作图
一些活性高的药物与相应受体结合的量效曲线 (B-log[L]曲线)并不一定与结合后产生效应的量效曲线(E-log[L]曲线)相重合。因为这类药物只需与一部分受体结合就能发挥最大效应(Emax),剩余下未结合的受体为储备受体(spare receptor)。这对理解拮抗药作用机制有重要意义,因为这类拮抗药必须在完全占领储备受体后才能发挥其拮抗效应。
受体激动药(L)对相应受体有较强的亲和力,也有较强的内在活性,α达100%。受体拮抗药(I)虽然也有较强的亲和力,但缺乏内在活性,α=0,本身不能引起效应,却占据一定量受体,拮抗激动药的作用。竞争性拮抗药(competitiveantagonist)能与激动药互相竞争与受体结合,这种结合是可逆性的。在实验中如果L与I同时存在则[RT]=[R]+[LR]+[IR],代入上述基本公式并加推导得
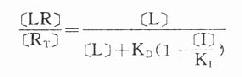
可见L和I同时存在时,如L这一因素固定不变,药理效应大小取决于[I]/K1(K1是I的解离常数)。[I]越高及(或)K1越小时效应越弱,即拮抗效果越强。当[L]>>[I]时,[LR]/[RT]→100%,这就是竞争性拮抗药使量效曲线平行右移(Emax不变)的理论解释(图2-9)。
在有一定量的竞争性拮抗药[I]存在时,增加[L]至[L’]仍可使药理效应维持在原来单用[L]时的水平。据此,
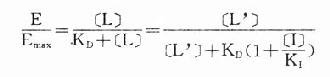
将之推导得
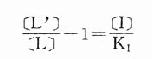
[L’]/[L]是剂量比 (dose ratio),即将[L]增加[L’]/[L]倍就能克服[I]的拮抗作用。该比值也取决于[I]/K1而与[L]绝对值或KD无关。将此公式两侧取log,并以log([L’]/[L]-1)为纵座标、以-log[I]为横座标作图,呈直线,斜率为1,与横座标交点为-logK1,即pA2此即Schild 图(图2-10)。按Schild定义,拮抗参数pAx是指剂量比为X时竞争性拮抗药浓度的负对数值。常用pA2,即[L’]/[L]=2时的数值,则pA2=-log[I]=-logK1,些参数反映拮抗药的拮抗强度,其值越大表示拮抗作用越强。
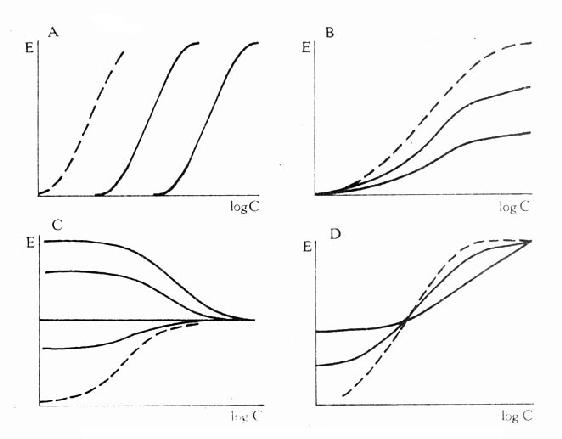
图2-9 竞争性拮抗药(A图)、非竞争性拮抗药(B图)及部分
激动药(D图)对激动药(虚线)量效的影响及激动药(C图)
对部分激动药(虚线)量效曲线的影响
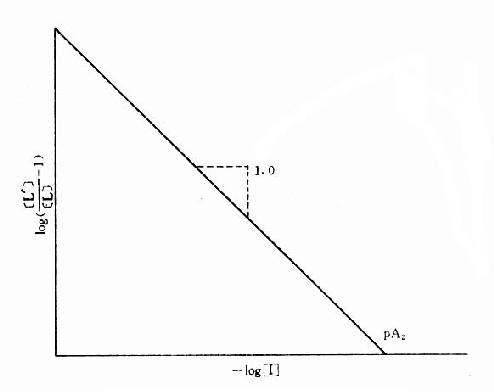
图2-10 竞争性拮抗作用的Schild作图
非竞争性拮抗药(noncompetitiveantagonist)与R结合非常牢固,分解很慢或是不可逆转,使能与L结合的R数量减少。另一类非竞争性拮抗药可阻断受体后某一中介反应环节而使受体-效应功能容量减少。二者共同特点是使量效曲线高度(Emax)下降。但L与剩余的R结合动力学不变,即KD不变。在双倒数图中更易看出这一关系(图2-11)。
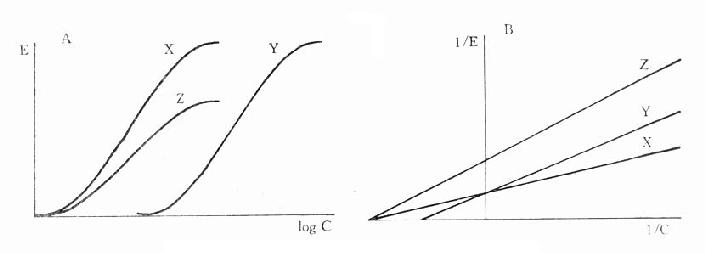
图2-11 竞争性拮抗作用与非竞争性拮抗作用比较
A图 量效曲线 B图 双倒数曲线
X 单用激动药 Y 竞争性拮抗药对激动药的拮抗作用
Z 非竞争性拮抗药对激动药的拮抗作用
还有一类药物称为部分激动药(partialagonist)和R结合的亲和力不小,但内在活性有限,α<100%,量效曲线高度(Emax)较低。与激动药同时存在时,当其浓度尚未达到Emax时,其效应与激动药协同,超过此限时则因与激动药竞争R而呈拮抗关系,此时激动药必需增大浓度方可达到其最大效能。可见部分激动药具有激动药与拮抗药两重特性。(图2-9C、D)
目前放射性配体-受体结合技术已普遍用于受体研究,但必需和药理效应实验结合进行才有意义。
为什么化学结构类似的药物作用于同一受体有的是激动药,有的是拮抗药,还有的是部分拮抗药?还可用二态模型(two-statemodel) 学说解释。按此学说,受体蛋白有两种可以互变的构型状态:静息状态(R)与活动状态(R*)(图2-12)。静息时平衡趋向R。活动药只与R*有较大亲和力,L-R*结合后充分发挥药理效应。部分激动药(P)与R及R*都能结合但对R*的亲和力大于对R的亲和力,故只有部分受体被激活而发挥较小的药理效应。拮抗药对R及R*亲和力相等,且能牢固结合,但保持静息状态时两种受体状态平衡,拮抗药不能激活受体但能阻断激动药作用。个别药物(如苯二氮
| 上一页:第四节 药物与受体 |
| 当前页:一、受体动力学 |
| 下一页:二、受体类型 |