<!DOCTYPE HTML PUBLIC "-//IETF//DTD HTML//EN">
Electrochemistry Software
ELECTROCHEMIST.com 5.8
Electrochemical
simulation and data analysis
DrHuang.com
226 Anzac Pde, Kensington, Sydney, NSW 2032, Australia
Mobile Phone: +61413008019
Fax: (61 2) 9662 0516
www.electrochemistrySoftware.com
Copyright @ 1990-2015
December 6, 2014
Contents
Chapter 2
Polarography and Voltammetry
2.2 Direct
Current Polarography
2.3 Linear
Sweep Voltammetry and Cyclic Voltammetry
2.5 Differential
Staircase Voltammetry
2.6 Alternating
Current Voltammetry
2.8 Additive Square Wave Voltammetry
2.10 Reverse Pulse Voltammetry
2.11 Differential
Pulse Voltammetry
2.12 Pseudo-Derivative
Normal Pulse Voltammetry
6.1 Simulating over
30 Factors
6.1.2
Effect of Hydrogen Ion Number
6.1.3
Effect of Reactant and Product Numbers
6.1.4
Effect of Electron Number
6.1.5.1
Effect of Electrode Geometry
6.1.5.2
Effect of Microelectrode
6.1.5.3
Effect of Electrode Area
6.1.5.4
Effect of Electrode Rotating Speed
6.1.5.5
Effect of Thin Film or Surface
Modified Electrode
6.1.6.2
Effect of Scan Direction
6.1.7
Effect of Preconcentration
6.1.7.1
Effect of Preconcentration Time
6.1.7.2
Effect of Preconcentration Potential
6.1.9.1
Effect of Pulse Height
6.1.10
Effect of Sampling Time
6.1.12
Effect of Diffusion Coefficient
6.1.13
Effect of Catalytic Reaction
6.1.13.1
Effect of Catalytic Reaction Rate
6.1.13.2
Effect of Concentration of Catalyst
6.1.15
Effect of Chemical Reaction Rate
6.1.16
Effect of Heterogeneous Standard Rate.
6.1.17.1
Effect of Adsorption Coefficient
6.1.17.2
Effect of Concentration
6.1.18
Effect of Electron Transfer Coefficient
6.1.22
Effect of Double Layer Capacitance
6.5
Calculating Theoretical Limiting Current
6.6
Extracting Parameters by Curve Fitting.
6.6.1
Fitting to Simulation Curve
6.6.2
Fitting to Experimental Curve
6.7 Separating Overlapped Peaks
6.8 Separating Faradic Current From Background
Current
Chapter 7 How Do
You Know It Is Right?
Chapter 8 Frequently Asked Questions (FAQ)
<big>Chapter 1
Introduction</big>
Software ELECTROCHEMIST.com (former Polar and Polarograph)
is virtual electrochemist who
can analyse and simulate electrochemical experiments. It simulates
- analytically and
digitally
- voltammetry and
chronoamperometry
- on virtually any
mechanism
- in 4 models (finite and
semi-infinite diffusions, convection and adsorption)
- at over 10 electrode
geometries (planar, spherical, semi-spherical, cylindrical,
semi-cylindrical, microdisc, and thin film electrodes, and their rotating
electrodes)
- by virtually any
waveform techniques (e.g. linear sweep, CV, DC, normal pulse,
differential pulse, square wave, additive square wave, staircase voltammetry,
user-defined waveform chronoamperometry and voltammetry).
It also simulates the effects to
change over 30 parameters, e.g. charge current, resistance, noise,
preconcentration time and potential, convection, pH, the reactant and
product numbers, etc. This
software provides five ways to check accuracy of simulation. It calculates
over 1000 types of theoretical peak values.
It plots and analyses any x-y data for peak location, peak height, peak width,
semi-derivative, semi-integral, derivative, integral, convolution,
deconvolution, curve fitting, and separating overlapped peaks and background
current.
It shows tip when the user put mouse cursor over a label. The program can separate overlapped voltammograms into individuals, and extract real peak from voltammogram with noise and baseline. It outputs the theoretical peak values, the peak current and potential and current-potential data, which can be imported into other program (e.g. Spreadsheet). Users can copy-and-paste the voltammogram into his document.
It has been successfully applied to fit experimental polarograms (voltammograms) of In(III), Cd(II), Pb(II), Tl(I), Cr(III), Zn(II), and binuclear copper complex in aqueous and non-aqueous media at mercury, solid metal and non-metal electrodes (specifically the dropping mercury, hanging mercury drop, gold, platinum and glassy carbon electrodes) by various electrochemical techniques (differential pulse, square wave, and pseudo-derivative normal pulse polarographies) [1-5].
It is available from the author or my Web site. If you have any question, please read FAQ in its document. For tutorial, please read the course practices in the rmit.htm file.
It is assumed that you agree the Shareware license that you should register by $10 to author in 20 days or you should delete it.
<big>Chapter 2
Polarography and Voltammetry</big>
2.1 Introduction
Modern
electrochemical methods offer the analytical chemist a wide variety of
techniques to solve analytical problems. Voltammetry is one such method, in
which the current is measured as a function of applied potential. Polarography
is another method, which differs from voltammetry in that it employs a dropping
mercury electrode (DME) to continuously renew the electrode surface.
In
this chapter, the fundamental principles of popular electrochemical techniques,
e.g. direct current
polarography (DCP), alternating current polarography (ACP), square wave
polarography (SWP), normal pulse polarography (NPP), differential pulse
polarography (DPP), pseudo-derivative normal pulse polarography (PDNPP), Linear sweep voltammetry (LSV) and
stripping voltammetry (SV),
are reviewed. Much of this theory is also applicable to voltammetry. If you are
familiar with polarography and voltammetry, they can move directly to the next
chapter.
Beside
techniques, theoretical equations also depend on mechanism and electrode
geometry. E.g. for 8 techniques, 15 mechanisms and 10 electrode geometries, we
need 8x15x10=1200 theoretical
equations. There are more than 1000 theoretical
equations for analytical simulation and theoretical peak current
and potential in this software. You
can calculate the peak (or limiting)
current and potential from
the theoretical equations by clicking on the
2.2 Direct Current
Polarography
Heyrovsky
invented the original polarographic method, conventional direct current
polarography (DCP), and Heyrovsky and Shikata constructed the first polarograph
in 1925 [6]. DCP involves the measurement of current flowing through the
dropping mercury electrode (DME) as a function of applied potential. Under the
influence of gravity, mercury drops grow from the end of a fine glass capillary
until they detach. Then the process is allowed to repeat itself. Drops may be
allowed to fall naturally or may be dislodged after a specified interval with
the aid of a mechanical device. A major advantage of the DME is that a
constantly renewed electrode surface is exposed to the test solution so that
problems of electrode blockage are avoided. Another advantage of the DME is
that it allows a number of electrode reduction processes to be monitored, which
would otherwise be inaccessible, because a wide negative potential region is
available on account of the high overpotential for water reduction.
If an electroactive species is capable
of undergoing a redox process at the DME, then an S-shaped current-potential
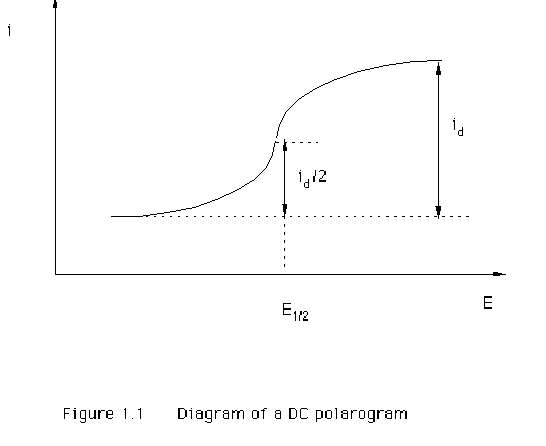
relation is usually observed. This is called a polarographic wave. Figure 1.1
illustrates the response obtained from a reduction reaction where the current
(i) increases over a particular potential (E) range until it reaches a limiting
value. The limiting current is the diffusion-controlled limiting current (id). This id is of interest in analytical
measurements as it is proportional to the concentration of reactant. For a
charge reaction
A + ne = B
Ilkovic [4] first put the measurement of this current on a theoretical basis, and his equation is [4-6]
id = (7/3)1/2 (36 p)1/6 r2/3 nF D1/2 m2/3 td1/6 C (2.1)
where r is the density
of mercury, n is the number of electrons, F is Faraday's constant, D is the diffusion coefficient, m
is the flow rate of mercury, td
is the drop time, and C is the concentration of the electroactive species in
the bulk solution.
For
a planar electrode,
id = nFAD1/2
C / (p
td ) 1/2
(2.2)
For
a spherical electrode with radius r,
id = id(planar)+nFADC / r = nFAD1/2 C/(p td )1/2 (1+ (p td D)1/2 / r ) (2.3)
For
a microelectrode, a steady-state current is
id = GnFA1/2 DC (2.4)
where G is an electrode geometry
constant, only depending on electrode geometry.
For
a microdisc electrode, G=4/(p) ½
id =4/(p) ½ nFA1/2 DC = 4nFDC r
For
a microsphere electrode, G=2p ½
id = 2p ½ nFA1/2 DC = 4pnFDC r
For
a microhemisphere electrode, G=p ½
id = p ½ nFA1/2 DC = 2pnFDC r
The
half-wave potential E1/2 is another important parameter of the
DC polarogram. This is the potential at which the current reaches half of its
limiting value (Figure 1.1). The value of half-wave potential is usually
independent of concentration and is characteristic of the electroactive
species. Therefore it can be used for qualitative characterization of the
species, and is the foundation of qualitative analysis.
The
shape of the DC polarogram is also very important to the overall
characterization of the electrode process. If the reduction reaction is
reversible and controlled by diffusion, the potential (E) is related to the
concentrations of reactant and product by the Nernst equation [7]:
E = E° + (RT/nF) ln( CO(0)/CR(0) ) (2.5)
where E° is the standard redox
potential, R is a gas constant, T is temperature, CO(0) and CR(0) are the surface concentrations of
species Ox and Red, respectively. The shape of the DC polarographic wave is
then derived by combining the Nernst and Ilkovic equations as follows [8,
9]
E = E1/2 + (RT/nF) ln( (id - i)/i )
or
i = id / [1 + exp( (nF/RT) (E - E1/2))] (2.6)
where
E1/2 = E° + (RT/2nF) ln( DR/DO
) (2.7)
Since
the diffusion coefficients of oxidized and reduced forms, DO and DR, are often almost equal, then E1/2 = E°. When i = id /2, then E = E1/2.
Equation
(2.6) is the Heyrovsky-Ilkovic equation, and is often used in investigations
into the nature of electrode processes. However, an experimental DC polarogram
also shows the oscillatory behavior of the current due to the growth and fall
of the mercury drop, and this is superimposed on the DC behaviour. This
invariably causes problems in the measurement of wave heights and/or half-wave
potentials, and of course has deleterious effects on measures of analytical
performance, especially sensitivity and resolution. Despite these problems, the
DME remains popular because of its constantly renewed surface.
2.3 Linear
Sweep Voltammetry and Cyclic Voltammetry
Linear
sweep voltammetry (LSV) is performed by applying a linear potential ramp in the
same manner as DCP. However, with LSV the potential scan rate is usually much
faster than with DCP. When the reduction potential of the analyte is
approached, the current begins to flow. The current increases in response to
the increasing potential. However, as the reduction proceeds, a diffusion layer
is formed and the rate of the electrode reduction becomes diffusion limited. At
this point the current slowly declines. The result is the asymmetric
peak-shaped I-E curve, as in Figure 1.3.
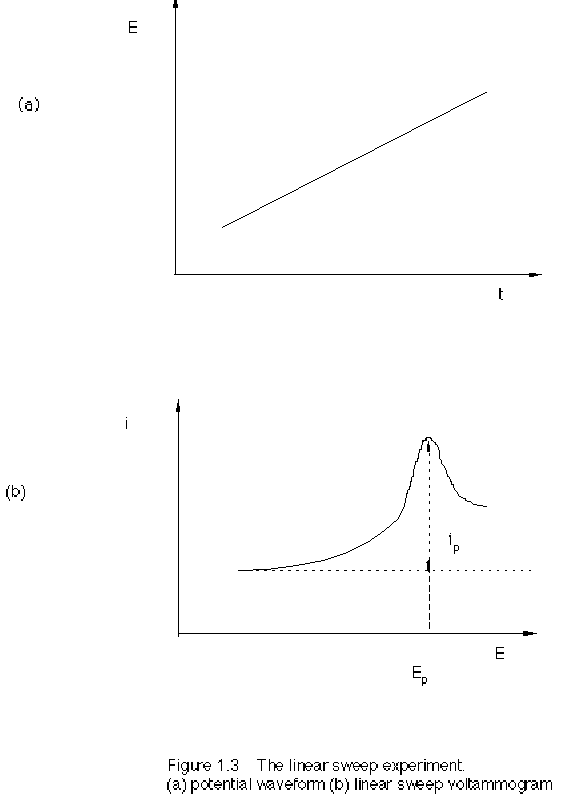
For a reversible reaction at a planar electrode, the peak current is
Ip = 0.4463 AC (nF) 3/2 (vD/(RT))1/2 (2.8)
The peak potential is
Ep = E1/2 – 1.109 RT/(nF) = E1/2 – 28.5/n (mV) at 25 °C (2.9)
The half-peak potential is
Ep/2 = E1/2 + 1.09 RT/(nF) (2.10)
The difference between peak potential and half-peak potential, similar to the half-peak width, is
| Ep - Ep/2 | = 2.2 RT/(nF) = 56.5/n (mV) at 25 °C (2.11)
Cyclic voltammetry is similar to linear sweep voltammetry except for the potential scans from the starting potential to the end potential, then reverse from the end potential back to the starting potential. Cyclic voltammetry is perhaps the most widely used electrochemical technique, and is frequently used for the characterization of a redox system. It can provide information about the number of redox states, as well as qualitative information about the stability of these oxidation states and the electron transfer kinetics. There are also simple models that can be used to calculate the rate of electron transfer (represented by ks) and the rate of chemical reactions coupled to the electron transfer for simple systems (those where the cyclic voltammetric behavior is controlled by only one of these parameters). However, these simple models cannot be used for more complicated systems, since the effects of, for example, slow electron transfer kinetics and a coupled chemical reaction cannot be readily separated. This simulation software can help quantitative studies (e.g., mechanistic investigations) in cyclic voltammetry, so it can be useful for investigating the electrochemical mechanisms of real redox systems. The difference between two peak potentials is
DEp =| Epa - Epc | = 2.3 RT/(nF) = 58/n (mV) at 25 °C (2.12)
E1/2 = (Epa + Epc
)/2
For a non-reversible reaction, DEp becomes larger.
For
a microdisk electrode, its steady-state current is the same as the eq. (2.4). Cyclic voltammetric responses at
a disk microelectrode can be approximated in simulation by using a hemispherical
electrode of the appropriate radius rh=2rd/p, where
rd is the radius of the disk microelectrode; the CV responses at a band
electrode can be approximated using a hemicylindrical electrode of the appropriate
radius rh=w/4, where w is width of the band electrode.
The rotating-disk electrode was
developed following the mathematical solution given by Levich of the
hydrodynamic equations describing the rate of transfer of substance in solution
to a rotating disk surface, in terms of the angular velocity of rotation ![]() (
(![]() , N
in rps), the diffusion coefficient D, the concentration C0 of the
substance and the kinematical viscosity
, N
in rps), the diffusion coefficient D, the concentration C0 of the
substance and the kinematical viscosity ![]() of the solution. For the case when the
reaction is relatively fast and the current is determined by mass transport,
the corresponding equation for the limiting current density
of the solution. For the case when the
reaction is relatively fast and the current is determined by mass transport,
the corresponding equation for the limiting current density ![]() ,
developed by Levich, is:
,
developed by Levich, is:
![]()
where ![]() is in A/cm2, D in cm2/s,
is in A/cm2, D in cm2/s,
![]() in cm2/s ,
in cm2/s ,![]() in
rad/s and C0 in mol/cm3. In such a case the limiting
current is independent of potential over a wide range. This range of potential
is limited at one end by the reversible potential and a small overpotential
needed to drive even a very fast reaction to mass-transport limitation and at
the other end by another reaction which may take place, usually the evolution
or oxygen or hydrogen in aqueous solutions.
in
rad/s and C0 in mol/cm3. In such a case the limiting
current is independent of potential over a wide range. This range of potential
is limited at one end by the reversible potential and a small overpotential
needed to drive even a very fast reaction to mass-transport limitation and at
the other end by another reaction which may take place, usually the evolution
or oxygen or hydrogen in aqueous solutions.
2.4 Staircase Voltammetry
Staircase Voltammetry (SV) is
similar to linear scan voltammetry. It scans
by staircase potentials, instead of linear potential. When a potential step is
very small, it is the almost same as linear scan voltammetry. But you have
choice to change the sampling time.
2.5 Differential Staircase Voltammetry
Differential Staircase Voltammetry (DSV) is
similar to staircase voltammetry.
Two currents are sampled at the beginning and the end of the staircase. When the difference between the two
current samples is plotted as a function of the applied ramp voltage, a
peak-shaped current response is shown.
2.6 Alternating Current
Voltammetry
A
number of modifications to DCP have improved its analytical performance. One of
them is alternating current voltammetry (ACV). It is the result of superimposing a
small amplitude sinusoidal potential (DE) with a fixed frequency (w)
on a slowly scanning DC ramp, as (c) in Figure 1.2. The applied potential is
then given by summing the AC and DC components. Finally, the alternating
current (AC) is measured as a function of DC potential. In particular, the
amplitude of the AC current vs. the DC potential is plotted, as (g) in Figure
1.2. The current-potential (I-E) curve for a reversible reaction follows the
equation [6]
I
= n2F2 AC DE (wD)1/2 sech2
[(nF/2RT)(E - E1/2)] /(4RT) (2.13)
At
a peak, sech()=1, then the above equation reduces to
Ip
= n2F2 AC DE (wD)1/2/(4RT) (2.14)
It
may be deduced from this equation that the amplitude of the AC component of the
Faradic current (I) is peak-shaped. Moreover, the peak current is a linear
function of concentration and therefore may be used in analytical applications.
Like the half-wave potential E1/2 in DCP, the peak potential Ep in ACP is characteristic of the
electroactive species. Also, the half-peak width (i.e. the width of the peak at
half its height, W1/2) is [6]
W1/2 = 3.52 RT/(nF) = 90/n mV at 25 °C. (2.15)
2.7 Square Wave Voltammetry
Square
wave voltammetry (SWV)
uses a small amplitude square wave voltage in place of the sinusoidal one used
in ACP. Its potential waveform is shown in (d) of Figure 1.2. The current is
sampled near the end of each square wave half cycle, to minimize double-layer
charging effects, and the I-E response is obtained by plotting the differences
in current between successive half cycles. For reversible electrode processes,
the I-E curve for SWP is similar to that in ACP [6], so its properties,
including the half-peak width W1/2 and resolution, are obviously akin to ACP.
2.8 Additive Square
Additive square wave polarography (ASWP) uses a small amplitude square
wave voltage in the same as
one used in SWP, but its
total current is sum of the positive and negative pulses currents, instead of
difference of the positive and negative pulses currents. Because its two charge
currents by the positive and negative pulses are opposite, it is possible to
select suitable sample time to make its charge currents offset to zero. A charge current by a positive pulse is
Ic(t1)
= (Ej-1 – Ej)exp(-t1/RC)= -(Es+Ep)exp(-t1/RC)
where t1 is
a sampling time at a positive pulse, R is resistance, C is double
layer capacitance, Es is potential step, Ep is pulse potential.
A charge current
by a negative pulse is
Ic(t2)
= (Ej – Ej+1)exp(-t2/RC)= Ep exp(-t2/RC)
Total
charge current is
Ic
= Ic (t1)+ Ic (t2)
By setting
Ic =0, a solution for the sampling time is
t2 = t1 -
RC ln(Es/Ep+1)
It can show
in dimensionless sampling time by division of the pulse time tp:
T2 = T1 –
RC/tp ln(Es/Ep+1)
According
to this equation, select sampling time t2 different from t1 to offset charge
current to zero.
2.9 Normal Pulse Voltammetry
The
pulse voltammetry including normal pulse voltammetry (NPV) and differential pulse voltammetry
(DPV) stem from
Barker's original work on square wave voltammetry [6]. The increased
sensitivity of these techniques over DCP arises from their ability to
discriminate against the charging current by measuring the total current after
the charging current has decayed to values substantially less than the Faradic
current.
The
potential-time waveform used in NPP is presented as (a) in Figure 1.2. At the
beginning of the potential sweep, the electrode is held at an initial potential
where no Faradic current flows.
Potential pulses of increasing amplitude are then applied to the
electrode at regular intervals. The potential pulses are about 50 ms in
duration and the current is measured at a time near the end of each pulse. A
potential pulse is ended by a return to the initial potential and the drop is
dislodged. The whole process is repeated except a few millivolts are added to
the potential pulse in next cycle. A normal pulse polarogram is shown as (e) of
Figure 1.2. The shape of the normal
pulse polarogram is sigmoidal, looking similar to the shape of a DC polarogram,
and indeed it can be described by a current-potential equation similar to that
in DCP [6].
For
a planar electrode, its limiting current is similar to one of DCV:
id = nFAD1/2
C / (p
tp ) 1/2
(2.16)
For
a spherical electrode with radius r,
id = id(planar) + nFADC / r = nFAD1/2 C (1/ (p tp ) 1/2 + D1/2 /r ) (2.17)
2.10 Reverse Pulse Voltammetry
Reverse Pulse Voltammetry is similar
to normal pulse voltammetry, but its start potential is negative and its pulse
is positive as opposite to normal pulse voltammetry.
2.11 Differential Pulse Voltammetry
Normal
pulse voltammetry gives improved sensitivity by avoiding most of the charging
current by sampling the total current as late as possible after the application
of each potential pulse. However, there still is the charging current to some
extent. Another defect of NPP is poor resolution between neighbouring wave
because of drawn-out sigmoidal I-E response. Differential pulse polarography
(DPP) was designed to overcome these problems by arranging a charging current
of smaller magnitude, and by producing a peak-shaped I-E curve.
The
potential-time waveform used in DPV
is shown as (b) of Figure 1.2. A voltage ramp is applied to the electrode as in
the DCP, and a small amplitude potential pulse (DE) is added to the voltage
towards the end of each drop's life. Two currents are measured before applying
the pulse and at the end of the pulse. When the difference between the two current
samples is plotted as a function of the applied ramp voltage, a peak-shaped
current response is shown.
The
peak-shaped I-E curve allows polarographic responses in close proximity to each
other to be more clearly resolved than in either DCP or NPV. The I-E curve for all values of the
pulse amplitude is described by [6]
I
= nFAC (D/ p tp)1/2 P (s2-1)/[(s+P)(1+Ps)] (2.18)
where
s
= exp(nFDE/(2RT)) (2.19)
P = exp[(nF/(RT))(E - E1/2 + DE/2)] (2.20)
At
a peak, P=1, then the current equation reduces to
Ip
= nFAC (D/ p tp)1/2 (s -1)/(1+s) (2.21)
Ep = E1/2 - DE/2 (2.22)
The half-peak width is a very
important parameter in resolution. The half-peak width W1/2 is a function of the pulse amplitude
as follows [6]
W1/2 = 2RT/(nF) cosh-1[2 + cosh(nFDE/(2RT))] (2.23)
For large values of |DE| (say |DE| > 200/n mV), W1/2 approaches to |DE|, and for small values of |DE| (e.g. |DE| < 20/n mV), this equation
reduces to equation (2.15).
Unfortunately,
the above theoretical equations are derived by neglecting the DC effect in DPP,
and although this is not a problem when the ratio of the drop time to the pulse
time is larger than 50, the resulting distortion makes the theoretical
treatment complicated, especially for a non-reversible reaction.
2.12 Pseudo-Derivative Normal Pulse Voltammetry
DPV is a very sensitive electroanalytical
technique due to the effective discrimination against the charging current.
However, DPV has two problems
associated with the slowly increasing DC ramp. As the DC ramp progresses,
filming may occur on the surface of the electrode if species form insoluble
mercury compounds [6]. Since the characteristics of the electrode are changed
by such a film, the current may not correspond to the simple theory. Another
problem is that the theory itself is complicated by the effect of the DC ramp.
NPP avoids these two problems. But the disadvantage of NPV is its poor resolution because of the
sigmoidal wave. To overcome this shortcoming, NPV polarograms can be differentiated to
produce peak-shaped responses, and thus combine the best features of both DPV and NPV while avoiding some of their
limitations. This pseudo-derivative normal pulse polarography (PDNPV) nevertheless is not sensitive as DPV.
The
potential-time waveform in PDNPV
is as in NPV, but the current
data of PDNPV are displayed in
a difference mode. The current is subtracted from those for the following
pulses, and the difference is plotted as a function of potential, as in DPV.
The
theoretical treatment of PDNPV
is simple and easy. The reversible current-potential equation is similar to
that of DPV except for the
DC term [6]. The half-peak width or resolution is akin to that of DPV.
2.13 Stripping Voltammetry
Stripping
voltammetry involves three main steps: electrodeposition (preconcentration),
equilibration, and stripping. The first step is to concentrate the analyte from
the dilute test solution into or onto the electrode at negative reduction (or
positive oxidation) potentials, usually accompanied by stirring. The second
step is to leave the solution to settle down. The third step is then to strip
the preconcentrated analyte from the electrode back into the solution by using
one of the polarographic techniques described above. A major advantage of this
method is its extremely sensitivity, in the concentration range of 10-6
- 10-12 M. This is because the concentration of the analyte on the
electrode is 100-1000 times greater than that in the starting solution [6].
<big>Chapter 3
Specification
This software analytically and digitally simulates voltammetry and chronoamperometry on virtually any mechanism in 4 models at over 10 electrode geometries by virtual any waveform techniques, calculates their theoretical peak current and potential, retrieve parameters by curve fitting, and separate overlapped peaks and baseline. </big>
- Digital simulation
Flexible for any mechanism up to second-order chemical reaction and adsorption by virtual any waveform techniques. You can type your mechanism and chemical symbols, and design or import any waveform. An implicit finite difference algorithm is used. - Analytical simulation
No divergence problem in simulation. No overflow problem in simulation. Fast simulation. - Design or import any waveform
Linear sweep, CV, DC, normal pulse, reverse normal pulse, differential pulse, square wave, additive square wave, staircase voltammetry. Multi-cyclic voltammetry, cyclic normal pulse, cyclic differential pulse, cyclic square wave, cyclic additive square wave, cyclic staircase voltammetry, differential staircase voltammetry, chronoamperometry, chronocoulometry, user-design waveform chronoamperometry and voltammetry. - Chemical
Reaction Rate up to 10e300
- Surface concentrations
show what happen each species in the electrode surface, check accuracy of simulation, convert surface concentration to current or current to surface concentration.. - Conversion between surface concentration
and current
Convert surface concentration to current or current to surface concentration to check accuracy of simulation. - Over 1000 type of theoretical peak values
It calculates theoretical peak values by theoretical equations and compare your data with theoretical peak values to see if your experimental conditions reach upper or lower theoretical limit. - Over 30 factors
Simulate over 30 effect factors, e.g. noise, charge current, resistance, preconcentration time and potential, convection, pH, reactant number, product number, electron number, electrode geometries, electrode size, electrode rotating speed, scan rate, concentration, pulse height, pulse width, sampling time, scan direction, scan cycle, diffusion coefficient, drop time, standard redox potentials, rate of electron transfer, transfer coefficient, diffusion coefficient, temperature, electrode area, and experimental parameters, forward and reverse chemical reaction rate constants up to 10e300, etc.
- Separating overlapped peaks
Manually and auto separates overlapped peaks into individuals, and extract real peak from voltammogram with noise and baseline. So you can exactly determine peaks and check accuracy of simulation. - Preconcentration
Change preconcentration conditions for stripping voltammetry. - Pre-equilibration
Calculate the concentration at equilibrium.
- Curve fitting
Manually and auto fits the simulated voltammograms into experimental data, and extracts kinetic parameters from experimental data. Curve fitting to any electrochemical parameter is easy by click to select that parameter. - Import and export data
Import parameters, waveform and current data from a text file. Copy-and-paste the simulated voltammogram into your document. Export the simulated data into your favor program (e.g. MS Excel). - Data Analysis
Derivative, integral, semi-derivative, semi-integral, convolution, deconvolution, Tafel analysis, convolution analysis. Semi-derivative is useful for CV. It can change a shape of reversible CV into symmetric peak, so easy to determine peak.
- Over 10 electrode geometries
Planar, spherical, semi-spherical, cylindrical, semi-cylindrical, band, microdisk, thin film, disk, ring electrode, and rotating all these electrodes. - Check accuracy of simulation
Provide five ways to check accuracy of simulation.
- Tips
Show tips for help when you put mouse cursor over a label.
Table 1 Feature
|
Version |
Shareware |
Student |
Teacher |
Academics |
Professional |
Competitor |
|
Digital simulation |
y |
y |
y |
y |
y |
y |
|
Analytical simulation |
y |
y |
y |
y |
y |
n |
|
Theoretical peak |
y |
y |
y |
y |
y |
n |
|
No. of adsorption reaction |
1 |
2 |
3 |
4 |
5 |
n |
|
Surface concentration |
y |
y |
y |
y |
y |
n |
|
Convert surface concentration to current |
y |
y |
y |
y |
y |
n |
|
Convert current to surface concentration |
y |
y |
y |
y |
y |
n |
|
Any species symbol |
y |
y |
y |
y |
y |
n |
|
Tips |
y |
y |
y |
y |
y |
n |
|
Open parameter |
n |
n |
y |
y |
y |
n |
|
Open waveform |
n |
n |
y |
y |
y |
n |
|
Open current |
n |
n |
n |
y |
y |
y |
|
Save parameter |
n |
n |
n |
y |
y |
n |
|
Save waveform |
n |
n |
n |
n |
y |
n |
|
Save current |
n |
n |
n |
n |
y |
y |
|
Save graph |
y |
y |
y |
y |
y |
n |
|
Show pulse current |
n |
n |
y |
y |
y |
n |
|
No. Of charge reaction |
1 |
2 |
3 |
4 |
5 |
y |
|
No. Of chemical reaction |
0 |
2 |
4 |
6 |
9 |
y |
|
Techniques: |
|
|
|
|
|
|
|
LSV and CV |
y |
y |
y |
y |
y |
y |
|
DC |
n |
y |
y |
y |
y |
n |
|
Normal pulse |
n |
n |
y |
y |
y |
n |
|
Reverse Pulse |
n |
n |
y |
y |
y |
n |
|
Differential pulse |
n |
n |
y |
y |
y |
n |
|
Cyclic diff. pulse |
n |
n |
y |
y |
y |
n |
|
Square wave |
n |
n |
n |
y |
y |
n |
|
Cyclic square wave |
n |
n |
n |
y |
y |
n |
|
Additive square wave |
n |
n |
n |
y |
y |
n |
|
Staircase |
n |
n |
n |
y |
y |
n |
|
Potential step |
n |
n |
n |
n |
y |
n |
|
Design waveform |
n |
n |
n |
n |
y |
n |
|
Import waveform |
n |
n |
n |
n |
y |
n |
|
Effect: |
|
|
|
|
|
|
|
Adsorption |
y |
y |
y |
y |
y |
n |
|
Convection |
y |
y |
y |
y |
y |
n |
|
Noise |
y |
y |
y |
y |
y |
n |
|
Charge current |
y |
y |
y |
y |
y |
y |
|
Resistance |
y |
y |
y |
y |
y |
y |
|
Reactant number |
y |
y |
y |
y |
y |
n |
|
Product number |
y |
y |
y |
y |
y |
n |
|
Preconcentration time |
y |
y |
y |
y |
y |
n |
|
Preconcentration potential |
y |
y |
y |
y |
y |
n |
|
Pre-equilibration |
y |
y |
y |
y |
y |
y |
|
pH |
y |
y |
y |
y |
y |
n |
|
Negative electron number |
y |
y |
y |
y |
y |
n |
|
Fractal electron
number |
y |
y |
y |
y |
y |
n |
|
Pulse height |
n |
n |
y |
y |
y |
n |
|
Pulse width |
n |
n |
y |
y |
y |
n |
|
First sampling time |
n |
n |
y |
y |
y |
n |
|
Second sampling time |
n |
n |
y |
y |
y |
n |
|
Analysis: |
|
|
|
|
|
|
|
Differentiate |
y |
y |
y |
y |
y |
n |
|
Integrate |
y |
y |
y |
y |
y |
n |
|
Semi-differentiate |
y |
y |
y |
y |
y |
n |
|
Semi-integrate |
y |
y |
y |
y |
y |
n |
|
Manual fit |
n |
y |
y |
y |
y |
n |
|
Auto fit |
n |
n |
n |
y |
y |
y |
|
Manual separate |
n |
n |
n |
n |
y |
n |
|
Auto separate |
n |
n |
n |
n |
y |
n |
|
Electrode: |
|
|
|
|
|
|
|
Planar |
y |
y |
y |
y |
y |
y |
|
(Micro) spherical |
y |
y |
y |
y |
y |
y |
|
(Micro) hemispherical |
y |
y |
y |
y |
y |
y |
|
cylindrical |
y |
y |
y |
y |
y |
y |
|
semicylindrical |
y |
y |
y |
y |
y |
n |
|
DME |
y |
y |
y |
y |
y |
n |
|
Microdisc |
y |
y |
y |
y |
y |
n |
|
Band |
y |
y |
y |
y |
y |
n |
|
Thin film |
y |
y |
y |
y |
y |
n |
|
Ring |
y |
y |
y |
y |
y |
n |
|
Rotating all electrodes |
y |
y |
y |
y |
y |
n |
Note:
y = yes, n = no. Feature may be
changed without notice.
<big>Chapter 4
Menu</big>
File menu
· Open Parameter submenu
Open a file of parameters
and read parameters back. You can continue your simulation of last time or
repeat other people’s simulation. The
Plot window title will show the file name.
·
Open Waveform submenu
Read waveform data from a file
and show the waveform. The Plot
window title will show the file name. If technique is 9) Design waveform, then it writes a few design points back to the Design Waveform panel.
·
Open Current submenu
Read current data from a file
and show curves. The Plot window title will show the file name.
·
Save Parameter submenu
Save experimental parameters into a text file. You can continue your simulation
later by the Open Parameter submenu.
·
Save Waveform submenu
Save waveform to a file as text file. E.g. if you save data as the .csv
file, you can load it into MS Excel by double-clicking the .csv file. If
technique is 9) Design waveform,
then it saves a few design points from the Design
Waveform panel, otherwise it saves
every point of waveform.
·
Save Current submenu
Save current data as a text
file. E.g. if you save data as the .csv file, you can load it into MS
Excel by double-clicking the .csv file.
· Save Graph submenu
Save a graph of current, waveform, and surface concentration to a file as .bmp file. You can use the PAINT program to save it as .gif file to show on Internet.
- Copy To Clipboard submenu
Copy a graph (e.g. current, waveform, surface concentration) into clipboard, so you can paste the graph into your document. - Print submenu
Print graph (e.g. current, waveform, surface concentration). - Exit submenu
Input menu
- Technique submenu
Open a window to select one of 9 techniques. The default technique is LSV and CV.
- Instrument submenu
Open a window to change instrument parameters. You can click the Reset button to use the default values.
- Mechanism submenu
Open a window to input your mechanism and species symbol in Digital simulation, or choose a predefined mechanism in Analytical simulation. The default mechanism is Fe3+ + e = Fe2+.
- Kinetics submenu
Open a window to change kinetic parameters. You can use the default values without any change.
- Concentration submenu
Open a window to change concentration, diffusion coefficient, adsorption coefficient and maximum adsorption amount of species.
Run menu
- Simulate submenu
Run simulation, and show curves on a Plot window. You can click on any point of curve to get the x and y values. - Manual Fit submenu
Manually fit the simulated curve into experimental curve as you manually change parameter values. - Auto Fit submenu
Automatically fit the simulated curve into experimental data. - Manual Separate
submenu
Manually separate the overlapped peaks into individuals as you manually change parameter values. - Auto Separate
submenu
Automatically separate the overlapped peaks into individuals.
Plot menu
·
i vs. E submenu
Plot current i versus potential E without running simulation.
·
i s vs. E submenu
Plot the sampling currents versus potential E without running simulation. It is
only available for multi-sampling techniques, e.g. pulse.
·
C0 vs. E submenu
Plot surface concentration C0 versus potential E without running simulation.
·
Waveform submenu
Plot potential E versus time t, which is imposed to electrodes in a technique.
· Convert submenu
Convert current into the surface concentration or the surface concentration into current.
· Convert
i to C0 submenu
Convert current
into the surface concentration.
· Convert
Co0 and Cr0 to i submenu
Convert surface concentrations of both oxidized and reduced species into current.
· Convert
i1 to C0 for E mechanism 1 submenu
Convert current to surface concentration of oxidized and reduced species for simple charge reaction mechanism 1 in the Analytical Simulation panel.
· Convert Co0 to i1 for E mechanism 1 submenu
Convert a surface concentration of oxidized species into current for simple charge reaction mechanism 1 in the Analytical Simulation panel.
· Convert Cr0 to i1 for E mechanism 1 submenu
Convert a surface concentration reduced species into current for simple charge reaction mechanism 1 in the Analytical Simulation panel.
· Convert
i8 to C0 for E mechanism 8 submenu
Convert current to surface concentration of oxidized and reduced species for catalytic reaction mechanism 8 in the Analytical Simulation panel.
· Convert Co0 to i8 for E mechanism 8 submenu
Convert a surface concentration of oxidized species into current for catalytic reaction mechanism 8 in the Analytical Simulation panel.
· Convert Cr0 to i8 for E mechanism 8 submenu
Convert a surface concentration reduced species into current for catalytic reaction mechanism 8 in the Analytical Simulation panel.
· Semi- dy/dt submenu
Semi-differentiate the y
data with time t. Semi-differentiation is
the same as deconvolution of
current with 1/Ö(pt). Click twice this menu for two time semi-differentiation,
i.e. first order differentiation.
· Semi-integrate submenu
Semi-integrate the y data
with time t. Semi-integrate is the
same as convolution of current
with 1/Ö(pt). Click twice this menu for two time
semi-integration, i.e. integration.
· dy/dt submenu
Differentiate the y data
with time, dy/dt. Click twice this
menu for second order differentiation.
· Integrate submenu
Integrate the y data with time t.
· Smooth submenu
Smooth the y data.
· Log((i lim 1 - i)/(i – i lim 2)) submenu
Tafel plot.
It converts S-shape of curve to a linear line. E.g. it converts DC voltamogram,
convolution of CV into linear lines.
· X Data submenu
Operation on all X data of a curve, e.g. X + constant, X * constant.
·
t as X-axis submenu
Plot time t as X-axis.
·
E as X-axis submenu
Plot potential E as X-axis.
· 0.001X, 0.1X, 10X, 1000X submenus
Multiply 0.001, 0.1, 10, or 1000 on X data. If your experimental potential data is not in Volt unit, you should convert to Volt unit by this submenu.
· X data reverse submenu
Reverse the order of data.
· Y Data submenu
Operation on all Y data of a curve, e.g. addition and subtraction of curves, Y1 + Y2, Y2 – Y1, Y + constant, Y * constant.
-Y, 0.001Y, 0.1Y, 10Y,
1000Y submenus
Multiply –1, 0.001, 0.1, 10, or 1000 on Y data. If your experimental current data is not in Amp unit, you should convert to Amp unit by this submenu.
·
Option submenu
Change the plot options, color, line style, etc.
Analyze menu
Find the peak height, location, left and right sides half peak width, and
width of curves of the peak shape.
After open a window, you can copy the result by right click mouse.
- Find Halfwave E submenu
Find the half wave potential and limiting current of curves of the S shape.
Calculate the theoretical peak potential, peak current, left side half peak width, right side half
peak width and half peak width
from theoretical equations. Select a mechanism from Analytical Simulation in
the Mechanism window before you click
this menu. This submenu is active for Analytical
Simulation only.
·
Curve Number submenu
Show current curve number. So you can analyze this curve.
·
Next Curve Number submenu
Go to next curve number. So you can analyze this curve.
· Time submenu
Display the simulation time and curve-fitting results.
Help menu
- Logon submenu
Logon to activate menus by input of password. - Manual submenu
Display this manual. - Home Page submenu
- About submenu
Show version number and ID of this software.
Some menus will be activated only after you click the Simulate submenu or load data because they require data.
<big>Chapter 5
Input Menu
This menu has five submenus.</big>
5.1 Techniques Window
Basic Techniques:
1) Linear sweep and cyclic voltammetry and
chronoamperometry.
2) DC voltammetry and chronoamperometry.
3)
4) Differential pulse voltammetry and chronoamperometry.
5) Square wave voltammetry and chronoamperometry.
6) Additive square wave voltammetry and chronoamperometry.
7) Staircase voltammetry and chronoamperometry.
8) Potential step chronoamperometry: single, double, and
triple steps.
9) Design waveform
10) Import waveform
You can design your waveform in the Design Waveform section of the Instrument
window. You can import your waveform
data from a file by the Open menu of
the File menu.
You plot the time t as X-axis for chronoamperometry, and the
potential E as X-axis for voltammetry.
By data analysis, you can extend above techniques to more
techniques, e.g.
integrating chronoamperometric current leads to chronocoulometry, and
convoluting CV leads to convolution voltammetry.
Extended Techniques:
1. Chronocoulometry: by integrating
chronoamperometric current.
2. Convolution voltammetry: by convoluting
CV.
3. Differential, semi-differential and semi-integration voltammetry: by differencing, semi-differencing and semi-integrating CV.
4. Anode and cathode stripping
voltammetry.
5. Differential staircase voltammetry:
produce 2 staircase currents at
different sampling times in the same window with overlap, and then click the Y2-Y1 menu of the Y Data menu in the
Analyze menu.
6. Multi cyclic voltammetry of all
above voltammetry.
You can see their waveform that is applied to electrodes by click on the Waveform menu of the Plot menu.
5.2 Instrument Window
This Instrument window is used to define the parameters of the instrument in experiment as follows:
- The potential range of the experiment (E start, and E end).
- The scan rate (v).
- The number of potential scans: Cycles.
- Solution resistance (R u) and double layer capacitance (C d).
- The electrode Geometry (e.g., planar, spherical) and surface Area.
5.2.1 Instrumental Parameters panel:
E start: starting
potential (V).
E end: ending potential (V).
E step: step potential (V).
v: scan rate (V/s). For square wave voltammetry, v=E step/t pulse.
E pulse: pulse potential (V).
T: temperature (°C).
t pulse: pulse time or pulse width for pulse voltammetry (s).
t drop: mercury dropping time or pulse length in pulse voltammetry (s).
Noise: noise signal (A).
ts1: first dimensionless sampling time, value is
from 0.1 to 1. For square wave pulse, it is sampled in first pulse during of
first half square wave. For different pulse, it is sampled in during before
pulse. For normal pulse, it is sampled in pulse during. For staircase, it is
sampled in a staircase during. It is not used for LS and CV. For digital
simulation, you should set the Time Grid Factor in the Digital Simulation
Model section to about 10 before you change the sampling time less than 1.
ts2: second dimensionless sampling time, value
is from 0.1 to 1. For square wave pulse, it is for second opposite pulse of
second half square wave. For different pulse, it is sampled in pulse during. It
is not used for other techniques. For digital simulation, you should set the
Time Grid Factor in the Digital Simulation Model section to about 10
before you change the sampling time less than 1.
Scan:
Single: single scan.
Cycles: cyclic scan, e.g. cyclic
voltammetry (CV).
2 Cycles: 2-cycle scan.
3 Cycles: 3-cycle scan.
5.2.2 Electrode panel:
1. Planar: a
planar electrode.
2. (Micro) Spherical:
a spherical electrode or micro spherical electrode.
3. (Micro) Hemispherical:
a hemispherical electrode or micro hemispherical electrode.
4. Microdisk: a micro disc
electrode, radius < 1e-4 cm.
5. (Micro) Cylindrical:
a cylindrical electrode or micro cylindrical electrode.
6. (Micro) Hemi
cylindrical: a hemi cylindrical electrode or micro cylindrical electrode.
7. DME: a dropping mercury
electrode.
8. Ring-Disc:
ring-disc electrodes.
9. Thin film: a thin film electrode, surface modified electrode or thin layer cell, with finite diffusion.
10. Bond: a bond electrode.
11. Ring: a ring electrode.
All above
electrodes can be rotated.
Area: electrode
area (cm2). When you change the value of area, the value of radius
is changed automatically. The default value is 0.01.
Radius: electrode radius (cm). When you change the value of radius, the
value of area is changed automatically.
Length: electrode length for cylindrical electrode or micro cylindrical
electrode (cm).
Ring Radius 2: inner radius of ring electrode (cm).
Ring Radius 3: outer radius of ring electrode (cm).
Thickness: thickness of the polymer or mercury film electrode (cm).
Rotation: electrode rotation rate (rpm). For stationary electrodes, set this value to 0. The default value is 0.
5.2.3 Preconcentration panel:
E pre:
preconcentration potential (V).
R stir: stirring rate (rpm).
Stirring solution.
t pre: preconcentration time (s).
t pre const: preconcentration time
constant (/s).
5.2.4 Baseline panel:
C dl: double
layer capacitance for charge current (F).
R: resistance (Ohm).
I start: a starting current (A).
I end: an ending current (A).
5.2.5 Digital
Simulation Model panel:
Space Grid Factor: space expanding grid factor. Its value is
from 0.001 to 0.9. The smaller value it is, the more accuracy simulation is,
but the longer computer time. Default value is 0.5.
Time Grid or
Step: the time step in second.
The smaller value it is, the more accuracy
simulation is, but the longer computer time. It depends on techniques. The
suggestion value is half pulse width for normal pulse, staircase, differential
pulse, and square wave techniques.
This section
factors are used for digital simulation only, not for analytical simulation.
The most important three parameters are the Space grid factor, the Time Grid and the Potential steps, which specify the
resolution of the space and time grids, respectively, that are used in the
simulation. Entering lower values for the Expanding grid factor and the
Potential steps parameter or higher
value for the Time Grid Factor will increase the resolution of
the grid, which may increase the accuracy of the simulation. However, there is a point beyond which further increases in
resolution will have no effect. Increasing the grid resolution will also
increase the time required for the calculation, but this is generally no longer
an issue with the speed of PCs now available. There are two occasions when
decreasing the Space Expanding grid factor is useful, and these are discussed
in later chapter (see Chapter 7 How Do You
Know It is Right?).
5.2.6 Design Waveform panel:
You can design your waveform that is applied to electrodes by setting your time-potential values here. This section is only visible for the Technique 9) Design waveform.
t0 is the start time, t0=0 second; E0 is the start potential (V).
t1 is the first point of time to change potential; E1 is the first changed potential.
t2 is second point of time to change potential; E2 is the second changed potential
and so on.
You can see its waveform by click on the Waveform menu of the Plot menu.
Default value is double steps scan.
5.3 Mechanism Window
You can type in your mechanism in the Digital Simulation panel with any symbol. Upper case symbol is different from lower case symbol. In order to faster computation, you should type in reactants only without products if chemical reaction is irreversible.
Tick
the checkbox under Adsorb for adsorption reaction. The adsorptive system
assumes that the adsorption obeys Langmuir isotherm and all species can be adsorbed.
For non-adsorptive species, set
its adsorption coefficient value to 0.
Uncheck the Digital Simulation checkbox, you will see the Analytical
Simulation panel. In the Analytical
Simulation panel, you
choose a predefined mechanism. Although
the electron number and reactant and product numbers are inside the Digital
Simulation panel, they are for both Digital and Analytical Simulations. You
can input any value of the electron number (n), e.g. n = -0.5.
Some of about 20 predefined mechanisms are as follows:
A+ne = B charge reaction
A+ne <-> B reversible charge reaction
A(a)+ne = B(a) Langmuir adsorption reaction
A(a)+ne <-> B(a) reversible Langmuir adsorption reaction
5.4 Kinetics Window
It is used to enter thermodynamic and kinetic parameters for the reactions involved in the mechanism. The following must be defined for each (Heterogeneous) electron transfer reaction:
- The redox potential (E°).
- The transfer coefficient (a).
- The electron transfer rate
constant (ks).
Heterogeneous Reaction Section:
ks: heterogeneous
standard rate constant (cm/s).
a: electron transfer coefficient.
E°:
standard electrode potential (V).
Three parameters are required for
each chemical (Homogeneous) reaction: the equilibrium constant (Keq), and the rates of the
forward and reverse reaction (kf and kb). Only two parameters kf and kb can be defined by the user,
since Keq = kf/ kb.
Homogeneous Reaction Section:
kf: forward
chemical reaction rate constant. Its unit is /s for the first order reactions,
or /sM for second order reactions. Its value is up to 1e300.
kb: backward chemical reaction rate
constant. Its value is up to 1e300
Keq: chemical equilibrium constant,
Keq = kf / kb.
Solution Section:
Electrolyte: electrolyte in solution.
C: concentration of electrolyte (M).
pH: the pH value of solution. The default
value is 7.
Vs: volume of solution (ml).
5.5 Concentration Window
The Species Parameters are
entered in this dialog box. These are the diffusion coefficients (D) and
concentrations of all the species involved in the redox mechanism. Two
concentrations are shown here.
The user enter the analytical
concentrations (C anal), which
are corresponding to the bulk concentrations
that in the solution. The initial
concentrations (C init) are the equilibrium concentrations at the
electrode surface, and are determined by E start, all Eo values,
all Keq values, and all C anal values. It is the C init
values rather than the Canal values that are used in the simulation. The
calculation of the C init values can be switched off by disabling the Pre-Equilibration
in its checkbox. If the calculation of C init
is disabled, the Canal values are the same as C init.
Species panel:
C anal:
analytical concentration (M).
C init: initial concentration at equilibrium (M). This concentration is used for simulation
and theoretical calculation.
C fitted: fitted value of
concentration (M).
C min: minimum concentration for
fitting (M). The minimum value usually is 0.1 time initial value.
C max: maximum concentration for
fitting (M). The minimum value usually is 10-time initial value.
D: diffusion
coefficient (cm2/s). The default value is 10^-5.
D fitted: fitted value of diffusion
coefficient (cm2/s).
D min: minimum diffusion coefficient
(cm2/s) for fitting. The minimum value usually is 0.1 time initial
value.
D max: maximum diffusion coefficient
(cm2/s) for fitting. The minimum value usually is 10-time initial
value.
b: Adsorption coefficient (/M). The default values of all
species are 10^4. For non-adsorptive species, set its value to 0.
Gm: Maximum adsorption amount (mol/cm2). The default value is 10^-8.
Pre-equilibration checkbox:
When this option is enabled, it automatically assumes that all the chemical and electrochemical reactions in the vicinity of the electrode surface are in equilibrium as determined by the thermodynamic parameters: chemical equilibrium constant Keq, the standard potential E°, and by the starting electrode potential Estart. Then, the entered values of analytical concentrations are not identical to the corresponding initial concentrations.
It is a good idea to keep the pre-equilibration option enabled. When the pre-equilibrated and analytical concentrations are different significantly, the initial condition for the experiment and the simulation may not be what was expected. The degree, to which the pre-equilibrated concentrations may be considered to be the bulk concentrations, will depend upon time of pre-equilibration (i.e., the time between setting the starting potential and initiating the potential scan), the operative kinetics, and the geometry. The value of the initial concentrations will act as if they are the bulk concentrations. A reasonable assumption only if the electrode geometry does not produce steady-state diffusion and if the pre-equilibration time is much longer than the duration of experiment.
When the pre-equilibration is not selected, the pre-equilibrated and analytical concentrations are the same.
<big>Chapter 6
Playing Around</big>
6.1
Simulating over 30
Factors
A simplest way to run simulation is
just to click the Run menu and then the Simulate submenu. It uses
the default values to simulate a linear sweep voltammogram. You can change technique under the Technique menu, or
change mechanism in the Mechanism window under the Mechanism menu, or change instrumental parameters
in the Instrument windows under the Instrument menu, kinetic
parameters in the Kinetic window under the Kinetic menu, or
concentration and coefficients parameters in the Concentration window
under the Concentration menu.
You have choice for digital or analytical simulation by clicking the Digital
Simulation checkbox in the Mechanism window. The analytical simulation is fast, and
useful for comparison of digital simulation.
Notice that some menu (e.g. the Plot
menu and the Analyze
menu) will be activated only after run simulation or load data because they
require data.
This software can simulates the effects with changing over 30 factors, e.g. charge current, resistance, noise, preconcentration time, preconcentration potential, convection, pH, the reactant number, and product numbers, standard redox potentials, rate of electron transfer, transfer coefficient, concentration, diffusion coefficient, forward and reverse chemical reaction rate constants, temperature, electrode area, and experimental parameters, etc.
This software can simulate many
cases shown in the book: "Electrochemical Methods, Fundamentals and Applications",
by AJ Bard [6].
A simple reversible charge reduction
reaction at a planar electrode by cyclic linear sweep technique (CV) is assumed
here, otherwise stated.
6.1.1 Effect of pH
Click the Mechanism menu to open a Mechanism window, tick
the “pH effect” checkbox, change the number of H+ in the
charge reaction, and then click the OK button to close the Mechanism
window. Click the Kinetics menu to open a Kinetics window, change
the pH value in the Solution section, and then click the OK
button to close the window. Run the simulation. You should see the peaks shift
when pH is larger or less than 7. As the pH value increases, the peak shifts to
more negative potential. For a charge reaction
a A + h H+ + ne = b
B
where a is
the reactant number, b is the product numbers, h is the number of
H+, and n is the electron number. The relationship of the
peak position with the pH value usually is linear:
Ep = k1
- k2 pH
Where k1 and
k2 are constants. k2 depends on the electron number, the number
of H+, the numbers of reactant and product, and temperature. For
a=b, it becomes
Ep = k1-
RTh/nF pH
For a=b and h=n, it
becomes
Ep = k1-
0.059 pH
It shows that both
peaks in CV shift to 59 mV more negative potential per pH. These agree with the
theoretical equation (2.5).
6.1.2 Effect of Hydrogen
Ion Number
As the hydrogen ion number h
increases from 1 to 2, the peak shift increase from 60 to 120 mV more negative
potential per pH. These agree with the above theoretical equation.
6.1.3 Effect of Reactant
and Product Numbers
For a charge reaction
a A + ne = b B
where a is
the reactant number, b is the product number, and n is the
electron number. If you change the reactant and/or product number of charge reactions, you should see the current changes.
As the product number increases,
e.g. the reaction A+e=2B, its current becomes lower and broader, the peak
shifts about 10 mV more negative.
As the reactant number increases,
e.g. the reaction 2A+e=B, its shape is the same as above reaction, and its
location and half-peak width are the same as above reaction, but the current
height is half.
For the reaction 2A+e=2B, its peak becomes more lower and broader. It is
the same as the reaction A+0.5e=B, because the former reaction becomes to the latter
reaction by division of the former reaction by 2. Its peak current is
0.5^1.5=0.35 lower than the peak current in the one-electron reaction A+e=B, which
agrees with the theoretical value in the eq. (2.8). Its peak potential Ep= E1/2
- 0.06 V, which is agree with the theoretical eq. (2.9). This is double
of the peak movement to more negative in the one-electron reaction. Its
half-peak width |Ep/2 – Ep|=0.11 V, which agrees with the
theoretical value in the eq. (2.11). This is double of the half-peak width
0.055 V in the one-electron reaction.
For the charge reaction 2A+2e=2B, its current is the same as the current
for the reaction A+e=B. This agrees with the eq. (2.5).
6.1.4 Effect of Electron Number
If you change the electron number of charge reactions in Digital
Simulation section for both Digital and Analytical Simulation, you should
see that peak height increases
and peak width decreases as the electron number increases. For LS technique at a planar electrode, its
peak current increases, which agrees with the eq. (2.8), its peak potential
shifts to more negative, which agrees with the eq. (2.9), and its half peak
width decreases, which agrees with the eq. (2.11).
If you change sign of electron number to negative, then reactant A
becomes a reduced species, product B becomes an oxidized species, and
the reaction becomes oxidation.
You not only simulate the effect of negative electron number, but also simulate the effect of fraction of electron number. E.g. electron number is 0.5.
Run two simulations for A+0.5e=B and 2A+e=2B. They are the same, which agrees with theory, as reaction A+0.5e=B is the same as 2A+e=2B.
6.1.5 Effect of Electrode
6.1.5.1 Effect of Electrode Geometry
You can simulate the effects of over
10 electrode geometries. Currents at different electrode geometries are
different as their diffusion models are different. By keeping the same area of
the electrodes, the peak current at the cylindrical electrode is larger than
the peak current at the planar electrode. The peak current at the spherical
electrode is larger than the peak current at the cylindrical electrode. These
agree with theoretical equations.
Note that the planar electrode
geometry is not available for microelectrodes. For larger planar electrodes, there is the edge effect, but it has little effect on the
over-all performance because the edges are small compared to the area of the
plane. As the electrode gets smaller, the relative contribution of the
edge becomes more important. A very small electrode, relative to the
thickness of the diffusion layer, behaves as if it is all edge.
6.1.5.2 Effect of Microelectrode
Not only the electrode geometry has effects on shape of current, but also the electrode size does. When the electrode size is very small, e.g. electrode radius is 1e-4 cm, its current becomes the S-shape from the peak shape, and steady-state current at the spherical electrode in LS technique is 1.2e-9 A, which agrees with the eq. (2.4). The steady-state current at the micro disc electrode in LS technique is 3.86e-10 A, which agrees with the eq. (2.4).
A shape of linear scan voltammogram
at spherical electrodes is
changed from peak shape to S-shape. When the products of scan rate and radius,
v r > 10-5, the
shape is peak. When v r < 10-7,
the shape is wave. The steady-state
current is independence of the time factors, e.g. the scan rate, the electrode-rotating
rate, the pulse time, the drop time, or the sampling time.
6.1.5.3 Effect of Electrode Area
The peak current increases linearly
with the electrode area for planar electrodes, or with square of the electrode radius
for planar disk electrodes. But it increases linearly with square root of the electrode
area or with the electrode radius for microelectrodes, regardless of electrode geometry,
spherical or disk electrodes. This agrees with theoretical equations.
6.1.5.4 Effect of Electrode Rotating Speed
For the rotating electrodes, current increases as the electrode rotating speed increases, and the
limit current increases linearly with square root of the electrode rotating
speed, which agree with theoretical equations. When the ratio of
rotating speed to scan rate, w/v < 1, the shape is peak. When high-speed w/v > 103,
the shape becomes S-shape wave. If you set the rotation speed to 0, the current should become one
without rotation.
6.1.5.5 Effect of Thin Film or Surface Modified Electrode
For a thin film electrode, surface modified
electrode or thin layer cell, its current shape is peak-shape as it is finite diffusion. Its behaves
are similar to adsorption reaction at plane electrodes. Its peak current
increases linearly with scan rate.
6.1.6 Effect of
Scan
6.1.6.1 Effect of
Scan Rate
For LS and CV techniques at a planar
electrode in simple reversible and irreversible charge reactions, the peak
current increases linearly as square root of scan rate increases, which agree
with the eq. (2.8).
In reversible and irreversible adsorption reactions, the peak current
increases linearly with increasing scan rate. So adsorption current increases
more rapid than diffusion current. But in quasi-reversible reaction, these
relationships are not linear anymore.
In catalytic reaction, the limit current is independent of scan rate.
For reversible charge and adsorption reactions, the peak location and
the width at half peak are independent of scan rate. For irreversible charge
and adsorption reactions, the widths at half peak are still independent of scan
rate, but the peak locations are not. The reduction peak location shifts
linearly to more negative potential and the oxidization peak location shifts
linearly to more positive potential as log of scan rate increases. Therefore, the
separation between the reduction and oxidization peaks becomes larger as scan
rate increases. These agree with theoretical equations.
For square wave and additive square wave
techniques, the peak current increases linearly as square root of frequency increases.
At a microelectrode, the steady-state
currents are independent of the time factors (e.g. the scan rate, the
electrode-rotating rate, the drop time, the pulse time, or the sampling time)
for all LS, DC, and normal pulse techniques, which agree with the theoretical
equations.
6.1.6.2 Effect of Scan Direction
For a reduction reaction, the scan direction
is from positive to negative, i.e. the start potential is large than the ending
potential, so the current is positive.
For an oxidation reaction, the scan
direction is from negative to positive, i.e. the start potential is less than
the ending potential, so the current changes to negative.
6.1.6.3 Effect of Scan Cycle
The current in second cycle is different
from current in first cycle. But the current in third cycle is close to the
current in second cycle. So, third cycle is enough.
6.1.7 Effect of Preconcentration
For anode stripping voltammetry, set the preconcentration
potential -0.2/n V more to
species’ standard electrode
potential, the start potential of
sweep to –0.3 V and the end potential to 0.3 V.
For cathode stripping voltammetry, set the preconcentration
potential 0.2/n V more to
species’ standard electrode
potential, the start potential of
sweep to 0.3 V and the end potential to -0.3 V.
Select the Preconcentration checkbox in the Instrument window.
6.1.7.1 Effect of Preconcentration Time
Change
the preconcentration time in the t pre field. The preconcentration time
usually is a number of minutes. If you increase the preconcentration time, e.g. from 600 second to 1000 second, the peak
current increases, but the peak current will have a limit. If you set the
preconcentration time to 0, you should see that the current is the same as one
without preconcentration. You should enter your mercury
film thickness into the Length field in the Electrode section of
the Instrument
window if you use a planar mercury film electrode.
6.1.7.2 Effect of Preconcentration Potential
Select
the Preconcentration checkbox
in the Instrument window. Change the preconcentration
potential value in the E pre field. If you increase the preconcentration potential, e.g. from 0 to –0.3 V for the standard
electrode potential of
0.1 V, the peak current increases, but the peak current will have a limit. It reaches
the limit when the preconcentration potential value
usually is -0.2/n V to species’
standard electrode potential for
anode stripping or 0.2/n V for cathode stripping. E.g. you further increase the preconcentration
potential, e.g. from –0.3
to –0.4 V, the current will not increase anymore.
6.1.8 Effect of Concentration
For a simple charge reaction, as the
bulk concentration of reactant increases, the peak currents increase linearly, but
the peak locations do not change, which agrees with the theoretical equations.
But for adsorption reaction, the peak current increases linearly in
lower concentration, then increase slowly nonlinearly, finally reach a limit at
high concentration, which agrees with the theory because it reaches maximum adsorption.
6.1.9 Effect of Pulse
6.1.9.1 Effect of Pulse Height
In differential pulse and square
wave voltammetry, for small pulse, the peak currents increase linearly but
resolutions become poor with increasing pulse height, which agrees with the
theoretical equations. For large pulse, the increasing of the peak currents is
not linear anymore.
But for additive square wave voltammetry, the pulse height has a little
effect.
6.1.9.2 Effect of Pulse Width
In pulse voltammetry, as the pulse
width increases, the peak or limiting current decreases. This agrees with the
theoretical equations.
For normal pulse and different pulse, square
wave techniques, the limiting or peak current decreases linearly as square root
of pulse time increases, which agrees with the eq. (2.2).
6.1.10 Effect of Sampling Time
As the sampling time decreases, the peak or limiting current increases, but
the charge current increases as well. This agrees with the theoretical
equations. The sampling time usually is larger than or equal to 0.6. In Staircase voltammetry, the peak potentials
shift to positive potential as well. For additive square-wave voltammetry, you
can change the first sampling time different from the second sampling time to
offset charge current to zero. But the sampling time does not affect steady-state
current at the microelectrode.
6.1.11 Effect of Techniques
You can simulate virtually any waveform techniques in voltammetry and chronoamperometry. You can design or import your waveform. You see the waveform applied to electrodes by click on the E vs t menu of the Plot menu.
From current shape point of view, techniques in voltammetry can be
divided into three types:
1. The first type is S-shape. E.g. DC and normal pulse voltammograms, steady-state current.
2. The second type is peak shape. E.g. differential pulse and
square wave voltammograms. But there is effect of the DC term on differential
pulse voltammogram. When you click the
i s vs E menu, you will see these pulse current and DC current.
3. The third type is the peak tailor shape. E.g. LS, CV, additive square
wave, and staircase techniques. Their current shapes usually are the peak
tailor shape, but depend on scan rate, electrode geometry, electrode size,
reaction mechanism, etc. The pulse currents in square wave technique are the
same as the current in the staircase technique when pulses become zero, which agree with theory.
6.1.12 Effect of Diffusion Coefficient
The change in the ratio of diffusion coefficient DA/DB leads to the
potential shift. It shows that the peak potential shifts to more positive as
the ratio increases. This agrees with theoretical equation dE/d ln(DA/DB)
= RT/(2nF). However, the height of the reverse peak almost does not change, although
a very small change occurs because of the changing relative position of Eend
and Epeak.
When the ratio = 4, the surface concentration of product at end is
double of the surface concentration of reactant at the beginning. When the
ratio = 1/4, the surface concentration of product at end is half of the surface
concentration of reactant at the beginning.
6.1.13 Effect of Catalytic Reaction
For catalytic mechanism
A+e = B, C+B->A
Assume that its charge reaction is
reversible, chemical reaction is irreversible, the concentration of species C
is much larger than the concentration of species A, and chemical reaction rate
is very large.
The currents in LS, CV, staircase, and
additive square wave techniques become S-shape from peak-shape.
6.1.13.1 Effect of Catalytic Reaction Rate
The limiting current increases linearly with
square root of the chemical reaction rate, but is independence of the time
factors, e.g. the scan rate, the electrode rotating rate, the drop time, the
pulse time, or the sampling time. It is similar to the steady-state current.
This agrees with the theoretical equations. For digital simulation, if you set
both chemical reaction rates kf = 0 and kb = 0, it becomes to a simple charge
reaction without catalytic mechanism.
6.1.13.2 Effect of Concentration of Catalyst
For above catalytic reaction, the limiting
current increases linearly with square root of the concentration of catalyst species
C. This agrees with theory.
6.1.14 Effect of Drop Time
For DC, NPV and DPV techniques, the limiting
or peak currents decrease linearly as square root of the drop time increases, which
agrees with the eq. (2.2).
6.1.15 Effect of Chemical
Reaction Rate
For EC reactions A+e=B, B=C, a reverse peak decreases as the forward
chemical reaction rate kf increases. But the reverse peak comes back
as the backward chemical reaction rate kb increases to equal to or
larger than kf. You can change the rate up to 10^300.
6.1.16 Effect of Heterogeneous Standard Rate
If the heterogeneous standard rate
constant ks is very large, e.g. 10^4, then the charge reaction is reversible,
and the heterogeneous standard rate constant has not any effect.
If the heterogeneous standard rate
constant is very small, e.g. 10^-4, then the charge reaction is irreversible,
and the heterogeneous standard rate constant has effect on the peak position
only, as the standard potential. For
irreversible charge reaction, the reverse peak is lower than the reduction
peak, but for irreversible adsorptive reaction, both peaks are the same height
if the value of a is
0.5.
6.1.17 Effect of Adsorption
The adsorptive system assumes that all adsorptions obey Langmuir isotherm and all species can be adsorbed. For non-adsorptive species, set its adsorption coefficient value to 0.
For reversible adsorption reaction, the forward and reverse currents are symmetric peaks in the same location and same height. The reverse current looks like mirror of forward current.
For non-reversible adsorption reduction reaction, the forward and reverse currents are not symmetric peaks in the same location anymore. The forward current peak moves to negative direction, while its reserve current peak moves to positive direction. So the peak separation becomes larger as the rate ks becomes smaller. This agrees with theoretical equations.
Adsorption voltammogram is similar
to thin-layer or thin film electrode one as all they are surface reactions. .
In the book by Bard [6]:
Case 1. For the Section 12.5.2 "Cyclic Voltammetry:
Only Adsorbed O and R
Electroactive - Nernstian Reactions" in page
521 of the book, you just tick on the select
box "Adsorb" in the Mechanism window, and click on the RUN menu.
Case 2. For the section 12.5.3 "CV: Irreversible Reaction" in page
523 of the book, it is similar to above Case 1, but you should change kf to
very small value, e.g. 1e-3.
Case 3. For section 12.5.4 "CV: Both Dissolved and Adsorbed Species
Electroactive" in page 525 of the book, its mechanism can be
A + e = B
O + e = R (adsorption reaction)
A = O, K=1
Parameters are K=1,
E1=E2, adsorption coefficients
of species A and B are 0.
(a) Product (R) Strongly Adsorbed.
Change the adsorption coefficient of product R
to 1e6, the adsorption coefficient of reactant O to 1e4. You should see CV is similar to the
Figure 12.5.4 in the page 527
of the book.
As scan rate
increases, adsorption peak
becomes higher.
(b) Reactant (O) Strongly Adsorbed.
Change the adsorption coefficient of reactant O to 1e6,
the adsorption coefficient
of product R to 1e4,
(c) Reactant (O) Weakly Adsorbed.
Other parameters: adsorption
coefficient of species R = 0, adsorption
coefficient of species O = 1e3. As
scan rate increases, adsorption
peak becomes higher.
(d) Product (R) Weakly Adsorbed.
Other parameters: adsorption
coefficient of species O = 1e-3, adsorption
coefficient of species R = 1e3. As scan
rate increases, adsorption peak
becomes higher.
6.1.17.1 Effect of Adsorption Coefficient
For reversible adsorption reaction with equal adsorption coefficients of reactant and product, peak location is at the halt-wave potential and half-peak width is 90/n mV. For strong adsorption, the adsorption coefficient affects the peak location, but did not change peak high. When adsorption coefficient of product is larger than adsorption coefficient of reactant, then peaks move to positive direction. When adsorption coefficient of product is smaller than adsorption coefficient of reactant, then peaks move to negative direction.
For irreversible adsorption reduction reaction, adsorption coefficients affect both peak high and location.
You can set
its adsorption coefficient value to 0 if species is non-adsorptive.
6.1.17.2 Effect of Concentration
The concentrations do not affect the adsorption peak current when adsorption reaches its maximum.
6.1.17.3 Effect of Scan Rate
The strong adsorption current increases linearly with scan rate. So, the adsorption current increases more than the non-adsorption current with scan rate. But it is not linearly anymore for weak adsorption.
6.1.18 Effect of Electron Transfer Coefficient
The transfer coefficient does not
affect on any reversible reaction.
For irreversible charge reactions, as the value of the transfer
coefficient increases, the
reduction peak is higher and narrower,
and the oxidized peak is lower and
wider. These agree with
theory.
6.1.19 Effect of Frequency
For both square wave voltammetry and additive square wave voltammetry, the currents increase linearly with square root of frequency. These agree with theory.
6.1.20 Effect of Temperature
Current decreases, the positive peak shifts a little bit to negative, and the negative peak shift a little bit to positive as temperature increases. These agree with theory.
6.1.21 Effect of Resistance
As resistance increases, both forward and backward currents in CV decrease and the peak separation increases. These agree with theory.
6.1.22 Effect of Double Layer Capacitance
As the double layer capacitance increases, the baseline current increases. These agree with theory.
The combination of capacitance and resistance lead that baseline is not linear anymore because they induce a non-constant dE/dt.
6.2 Surface Concentration
After run simulation, click the Plot
menu, and then click the C0
vs E submenu to show surface concentrations. The concentrations at the electrode surface are useful for checking accuracy
of simulation.
For a reduction reaction A+e=B, the concentration of reactant decreases
and the concentration of product increases as potential moves to more negative
since scan. The concentration of reactant decreases to zero and the
concentration of product increases to the same as initial concentration of
reactant at the end of scan. Because all amount of species A becomes the same
amount of B at the end of scan. Their concentrations cross at the half wave
potential. These agree with the theoretical eq. (2.5).
For a reduction reaction A+e=2B, the concentration of reactant decreases
to zero and the concentration of product increases to double of initial
concentration of reactant at the end of scan, which agrees with theory because
one molecular of species A produces two molecular of species B.
For a reduction reaction 2A+e=B, the concentration of reactant decreases
to zero and the concentration of product increases to half of initial
concentration of reactant at the end of scan, which agrees with theory because
two molecules of species A produce one molecular of species B.
For EE reactions A+e=B, B+e=C, it is interesting to see the maximum
surface concentration of the species B is lower than the species A or C.
For EE reactions A+e=2B, B+e=2C, the
maximum surface concentration of the species C is double of the species B, and
the maximum concentration of the species B is double of the species A, which
agrees with theory, because one molecular of species A produces two molecules
of species B and two molecules of species B produces four molecules of species
C.
For EE reactions 2A+e=B, 2B+e=C, it is opposite to the above reaction.
For effect of diffusion coefficient, see chapter 6.1.12 Effect of Diffusion
Coefficient.
The surface concentrations look like
the same in reversible simple reaction, regardless of scan rate, electrode
size, electrode geometry, and techniques
if pulse height is zero, digital simulation, and analytical simulation. For NPV
and DPV, the surface concentrations move in the pulse potential. For square
wave and additive square wave techniques, the surface concentrations move in
half the pulse potential.
You also can check accuracy of
simulation by converting surface concentration to current or current to surface
concentration under the Convert submenu of the Plot menu.
6.3 Comparing Curves
After run first simulation, click
the Plot menu, and then the Option submenu. Select the Overlap
checkbox, and then run second
simulation. You can change color and
line styles for individual curves. This software can compare up to six curves.
6.4 Analyzing Data
This software can analyze the x-y data for peak location, peak height, peak width, convolution, deconvolution, semi-derivative, semi-integral, derivative, integral, curve fitting, and separating overlapped peaks and background current. Semi-derivative is useful for CV. It can change the asymmetric peak shape of CV into the symmetric peak for easy measurement.
6.5 Calculating Theoretical Limiting Current
Click the Analyze menu and
then the
6.6 Extracting Parameters by Curve Fitting
The difficult part of a voltammetric experiment is extracting the chemical information from the current-voltage curve. Apart from very simplistic analysis, the measured current cannot be directly interpreted. This software can extract the chemical information from the whole current-voltage curve. It helps to get parameter values and mechanisms. Curve fitting to any electrochemical parameter is easy by click to select that parameter, and then click on the Manual Fit or Auto Fit menu.
6.6.1 Fitting to Simulation Curve
In order to extract kinetic parameters, you can fit a simulation curve to another simulated or experimental curve. It can retrieve any of 30 parameters (e.g. concentration C, standard electrode potential E°, and the heterogeneous standard rate constant ks) from voltammogram by curve fitting. Select parameters that you want to fit, and input the initial, minimum and maximum values of the parameters. The minimum value usually is 0.1time initial value and maximum value usually is 10-time initial value. e.g. after run simulation with all default values, select a concentration, then change the C value from 1e-3 to 2e-3 in the Species section, click the Auto Fit menu. You will see the fitted value of 0.001 in the C fitted field next to the C text field. Notice that when you auto fit, you should not click on the OK button on the Chemicals window to close the Chemicals window, otherwise you will get the “Runtime error 6: overflow”. This bug is fixed since version 4.6.
You should manual fit before auto
fit. The manual fit shows how well your initial guesses values work. If it
diverged, you should change their initial values and/or minimum and maximum
values, and then try again. By the
manual fit, you must change the initial values every time of run.
6.6.2 Fitting to Experimental Curve
One
of the key functions of this software is a fitting routine that optimizes
selected simulation parameters to provide the best fit between the experimental
and simulated voltammograms. Data is text file formats without header. There
are a number of important points to note:
- The data must be arranged as potential-current
couples.
- The potential and current values within a data
couple must be separated using a comma.
- The potential step must be constant.
It
should also be stressed that the potential step (i.e., the difference between
adjacent potential values) must be constant throughout the data set. We have
observed that variation of the potential step value can cause considerable
problems with the fitting routine.
It is similar to fit the simulated curves. Click the File menu, the Open submenu, the Cuurent Data submenu to select
your data file. But you should input your experimental values of Estart,
Eend, Estep, etc. into the Experimental
section. This software requires
that data are in SI unit and first peak is positive value. If your experimental
data are not, please convert your experimental data to in SI unit. E.g. click the Analyze menu, and then the 0.001Y
submenu to convert current from mA to A. After the experimental data (text) files are selected and loaded
into this software, the mechanism and parameter values
are then entered, and the parameters to be varied are selected. A parameter of
start current in Baseline section should be zero. Once these have been
done, you can start fitting
operation by clicking the Fit
menu.
It is important to note that any
given voltammogram may be
accurately simulated by more than one mechanism and/or set of parameter values.
Experimental measurements should therefore be made over a wide range of
parameter values. The most common variables are scan
rate and technique, although
variation of concentration and/or temperature
can also be used. If one set of parameter values can provide a good match between
the experimental and simulated
voltammograms measured over a wide range of scan rates (and/or techniques), then this is good
evidence that these parameter values are correct. However, it does not prove
that the correct mechanism and parameter values have been selected. It is up to the user to determine
whether the selected mechanism and parameter values are chemically and
electrochemically reasonable (i.e., are they consistent with the results of
electrochemical studies on similar systems?). The sensitivity of the fit to
variations in the parameters values must also be investigated.
It
should note that for irreversible charge reactions, you cannot fit both the heterogeneous
standard rate constant and the standard electrode potential in the same time
because they become dependent each other, and they are coupled.
· For a simple reversible or nearly reversible redox reaction,
A + e = B, it is in principle reasonable to evaluate Eo and D (= DA = DB) from
a single CV. Data over a range of scan
rates will confirm if it is diffusion control or not.
· If the reaction is quasi
reversible it also might be possible to obtain meaningful values for ks and a from a single CV. Here again, wisdom
dictates that data from several CVs run over a range of scan rates be used to
evaluate ks and a.
· Parameter values may be coupled: Under certain circumstances
there may be an infinite number of combinations of values of two (or more)
parameters that effect identical (or virtually identical) fits, e.g.:
For a simple charge reaction, A + e = B, the values of Eo and the ratio
DA / DB are coupled. This is why it allows diffusion
coefficient values to be linked during fitting- unlinked there would be an
infinite number of combinations of Eo values and DA / DB
ratios that would effect identical CVs.
For a completely irreversible, A + e -> B, the values of Eo and ks
are coupled.
For an EC reaction, A + e = B with an effectively irreversible chemical
reaction B -> C by kf, and RT kf / F|v| > 1, the
values of Eo and kf are coupled.
The manifestation of coupling can be
· Non-convergence.
· Convergence to vastly different parameter values depending
upon the initially guessed values.
· Wide confidence limits on one or more of the coupled
parameters.
A seemingly
excellent fit of the experimental and simulated data does not warrant high
confidence in the value of any given parameter.
Suggestions for experiment:
· Obtain experimental data over a range of scan rates, and vary
the starting and reverse potentials.
· Run multiple cycles.
· Alter the chemical conditions, e.g. change of concentrations
of reactants, pH.
· Vary the electrode size.
· Use known values for parameters.
Suggestions for simulation:
· Change the initially guessed values of the optimized
parameters to confirm that the optimized values remain the same.
· If you suspect that an optimized parameter is playing a minor
role, de-select it, and explore the effect of set it at several different
values while the remaining parameters are optimized.
· Be aware of sources of systematic error, e.g., uncompensated
resistance, capacitance; edge effects; adventitious stirring.
· Attempt to optimize the minimum number of parameters required
for a reasonable fit.
6.7 Separating Overlapped Peaks
For multi charge reactions, overlapped
peaks are usually observed. There are errors in determination of peak height
and position in each reaction as the overlapped peaks. It is necessary to
separate overlapped peaks into individual peaks and check accuracy of
simulation. If you click the Manual Separate submenu under the Run
menu, you will see individual peaks. Click the
6.8 Separating Faradic Current From
Background Current
Because double layer capacitor and
resistance, there is background current such as charge current. This software
provides two ways to separate Faradic current from background current.
1. To simulate
current with background current, click the Input menu, the Instrument
submenu, change the value of Cd to 0.0001 and the value of
resistance R to 10000 in the Baseline section, and run simulation. You
should see current with baseline. When click the Manual Separate menu,
you should see third curve for the Faradic current without background current.
2. To simulate
background current, click the Input menu, the Instrument submenu,
change the value of Cd to 0.0001, the value of resistance R to 1000
in the Baseline section and the value of the concentration C to 0 in the
Concentration window, and run simulation. You should see background
current. Then, select the Overlap checkbox in the Option window,
change the value of the concentration C to 1e-3 in the Concentration
window, and run simulation. You should see second curve for current with
background current. Finally, click the Plot menu, the Y Data
submenu, and the Y2-Y1 submenu. You should see third curve for the
Faradic current without background current.
<big></big>Chapter 7
How Do You Know It Is Right?
Any simulation procedure has its stability and accuracy limitations.
This software provides five ways to check for accuracy of simulation:
1. The first approach is to compare peak values of simulated
voltammograms with theoretical values.
Uncheck the Digital Simulation
checkbox to change to Analytical Simulation, select your mechanism, and then
click the
2. The second method
is to compare digital simulation voltammograms
with analytical simulation voltammograms. E.g. in order to compare digital
simulation of adsorptive
reversible reaction with analytical
simulation of reversible adsorptive reaction and general adsorptive reaction, run digital simulation of adsorptive reaction, click the Overlap checkbox in the
Option menu of the Plot menu, uncheck the Digital Simulation checkbox to see
the Analytical Simulation, select the mechanism 15, run simulation, select the
mechanism 17, run simulation, you will see 3 curves overlap together.
3. The third approach is to change the computational parameters. The exponential time and space grids used by the implicit finite
difference computation are characterized by t and x. Although these parameters
are not defined explicitly in the user interface, changing the potential steps, and the space expanding grid factor in the
Instrument window respectively can alter their values. Decreasing the values of
these parameters almost always improves the accuracy of a given simulation, but
the computation time is also increased. This
software sets default values for these parameters that will produce
acceptable accuracy (e.g. better
than 0.5%) in most cases. However, there are instances where the particular set
of the used parameter values
causes computational problems. Decreasing the values of one or both of
these Model Parameters can eliminate this problem. It is possible to obtain a simulated
voltammogram that looks reasonable but is still inaccurate. It is good practice
to run any simulation using different values for the expanding grid factor and the potential steps
to check for accuracy. A significant
difference in the results indicates that the default values are inadequate for
accurate simulation. Because the smaller values of the potential step and/or
space expanding grid factor will effect a noticeably longer computation time,
we should use the possible largest values, which retain acceptable accuracy.
4. The fourth method is to check the
concentration at the electrode surface, and to convert surface concentration to
current or current to surface concentration. See Section 2.6 Surface
Concentration.
5. The fifth way is to check the
separated individual currents for multi charge reactions by the Separate
menu.
You can combine
any of these five ways to check for accuracy of simulation.
<big></big>Chapter 8
Frequently Asked Questions (FAQ)
Q: How much does
registration cost?
A: Its membership is from $10 per year. Shareware version $10 + $10 per function
(except $100 for the function of the import experimental data, and auto fit $70)
per year per copy. Some functions must be ordered together. Please state what
function you want when you order. E.g. for a full package of all functions is
$480: Basic (Shareware) version $10 + import data $100 + export data $10 + 10
techniques $100 + manual fit $10 + auto fit $70 + manual separate $10 + auto
separate $10 + open parameter file $10 + save parameter file $10 + 5 charge
reactions $50 + 9 chemical reactions $90 = $480. It is Australian dollar.
It is recommended
that you try shareware version on a computer where you want to work before you
buy. You cannot change hard disk drive where it was installed after password is
given.
Q: Which platforms can the program run on?
A: Its 32-bit version program
runs on IBM PC under Windows 95/98/NT/2000/XP/Vista,
while its 16-bit version program
runs under Windows 3/3.1/3.11/95/98/NT.
The 32-bit version needs Microsoft Visual Basic 6 runtime DLL files (e.g. msvbvm60.dll, comdlg32.ocx) in the same directory as the program or in the directory \windows\system for Windows 3.11 or 95, or in the directory \winnt\system32 for Windows NT.
The 16-bit version needs Microsoft Visual Basic 4 runtime DLL files (e.g. vb40016.dll and oc25.dll) in the same directory as Polar or in the directory \windows\system for Windows 3.1, or in the directory \winnt\system for Windows NT.
Q:
I cannot save a file.
A: You miss the Microsoft Visual Basic 6 runtime DLL file comdlg32.ocx or you did not register.
Q: Where can I download these dll?
A: Microsoft Visual Basic 6 runtime DLL files are from http://www.simtel.net/simtel.net/win95/dll.html, where msvbvm60.dll is inside simvb6-5.zip. Microsoft Visual Basic 4 16-bit runtime DLL files are from http://www.simtel.net/simtel.net/win3/dll.html.
Q: When I click the
Simulate menu, I got error: “No data”, or "Run-time error 13”, with the message: "Type mismatch".
A: I guess you are running it under non-English version of Windows. Please change language setting to English in the Regional Setting of the Control Panel, and restart Polar. Or try it under English version of Windows. Some non-English versions of Windows have problem to run English version program.
Q: Still have install
problem?
A: You should close all programs (include Office, Mail) before install the program. If you still have
problem, try to register file msvbvm60.dll by double click or type following
command in DOS:
Cd \windows\system
Regsvr32 msvbvm60.dll
then start Polar.
Q: Why are some menus
inactive?
A: Some menus will be activated only after you click the Simulate menu
or load data because they need data.
Q: I cannot see any
chemical reaction in Shareware version. Is this part of the program not
finished yet or is it only available in the registered version?
A: It is only available in the registered versions. You can
change chemical reaction rate kf up to 1025. The registered versions
simulate virtually any mechanisms.
Q: Does it include my
mechanism?
A: If your mechanism is missing, please send your requirement into
author. Author may add your mechanism into new version special for you.
Q: Can it fit data by
curve fitting?
A: Yes. Click to select a parameter that you want to fit, and then click
the Auto Fit menu.
Q: Can I change graph into
other program Spreadsheet Excel?
A: Yes. You export data in text file, and then read data into it.
Q: Some submenus
semi-derivative, semi-integral, derivative, and integral, seem to not work
sometime. How can I do?
A: You should first click the Next submenu under the Plot menu, then try
semi-derivative submenu.
Q: How can I get
registered version?
A: You will receive it if you pay author registration fee.
Q: What are differences
among Shareware, Student, Teacher, Academics and Professional versions?
A: The Shareware version is for try before you buy, the Student version
is for students, the Teacher version is for teachers, the Academic version is
for academics, and the Professional version is for professionals. Please see Table
1 Feature for details.
Q: When I run the SWV with
default conditions, it does not appear to give the correct curve. Why?
Because default conditions are for linear sweep and CV only. For
Q: Is it possible to click on a point and then have
displayed both the current and potential for the point?
A: Yes, since version 4.7.
Q: How to simulate
oxidation reaction?
A: change the scan potential to the Estart < the Eend in the Instrument window.
Q: When I click on the Auto Fit menu, I got “Runtime error 6: Overflow”. Why? How to fix it?
A: Because you close the Chemicals window. When you auto fit, you should not click on the OK button on the Chemicals window to close the Chemicals window, otherwise you will get the “Runtime error 6: overflow”. It has been fixed since version 4.6.
Q: What data format can be imported?
A: The x-y pairs of text data. Please see Chapter 6.6.2 Fitting to Experimental Curve.
Q: Is it licensed for user
or machine?
A: Software is like hardware. If you want to use different PC, you had to buy different machines. Can you just buy a single machine in order to use different PC? Many users can share one machine. It is the same as many users can share one copy of software. Therefore, the software license is for machine, not for user. One copy of software is for one machine. If you want to use software for different PC, you should buy more copies of software, and you will get discount.
Q: What happen when I
upgrade machine?
A: When you upgrade the hardware of machine, you can change motherboard, CPU, RAM, add hard disk, but it is suggested that you should keep your old hard disk and never format it, so your software ID does not change, it will still work. You can put old hard disk into external hard disk box with USB connected to new PC. Or you install it in USB disk, not in hard disk.
Q: Can I run a single
copy of this software in different PC?
A: Yes, if you install it in USB disk, you can plug the USB disk into any PC, and use the same password to login. It is portable.
Q: How does it compare to
competitors?
A: This software has
many advantages over competitors (see details on the feature table in Chapter
2 Features). Some
advantages are as follows:
1. Competitor only digitally simulates a single technique CV at 5 electrode geometries, while this software analytically and digitally simulates virtually any waveform technique at over 10 electrode geometries.
2. Competitor cannot design or import waveform while this software can.
3. Competitor cannot simulate adsorption while this software can.
4. Competitor cannot simulate very fast chemical reaction rate larger than 1e20, while this software can simulate chemical reaction rate up to 1e300.
5. Competitor cannot simulate reactions with reactant or product number, e.g. 2A+e=B, while this software can.
6. Competitor cannot simulate reactions with fraction of electron number, e.g. A+0.5e=B, while this software can.
7. Competitor cannot separate overlapped peaks, while this software can.
8. Competitor cannot simulate effect of pH, while this software can, and even simulates over 30 effect factors.
9. Competitor cannot calculate any theoretical value, while this software can calculate over 1000 types of theoretical peak values.
10. Competitor cannot analyze data, while this software can.
11. Competitor cannot check simulation accuracy, while this software can by five ways of checking.
12. You download and try this software free.
13. This software is more powerful but cheaper.
Q: I still have questions.
A: Please
post your questions to Electrochemistry Forum in website www.electrochemistry.net.
<big>Chapter 9
References</big>
- W. Huang, T. Henderson, A.M. Bond and K.B. Oldham, Curve fitting to resolve overlapping voltammetric peaks: model and examples, Anal. Chim. Acta, 1995, 304, 1-15.
- W. Huang and B. Hibbert, Computers & Chem., 1995, 19(4), 433.
- W. Huang and B. Hibbert, Computers & Chem., 1995, 19(4), 435.
- W. Huang and B. Hibbert, Polar 2.0 for Windows: simulator of voltammogram, Chem. in Aus., 1996, 131.
- J. Mo, P. Cai, W. Huang and F. Yun, Theory and application on multiple semidifferential electrochemical stripping analysis with thin mercury film formed in situ, Acta Chimica Sinica, 1984, 42(6), 556-561, [CA 101: 162712].
- A. J.
Bard and L. R. Faulkner, Electrochemical Methods, John Wiley & Sons,
- D. Britz, Digital Simulation in
Electrochemistry, Third Edition, Springer,
- Roy Lowry, Polar, Physical Science Educational Review, 2002, Nov., 3(2), 26-27, http://www.physsci.ltsn.ac.uk/Publications/Journal/journ3_2.pdf.
- J. Mo, W. Huang and R.J. Zhang, New Advances in convolution voltammetry (Review), J. Anal. Determining (Fenxiceshi Tongbao), 1985, 4(3), 1-8, CA 105: 163910.